
Breast Cancer Type Classification Using Machine Learning


Abstract
:1. Introduction
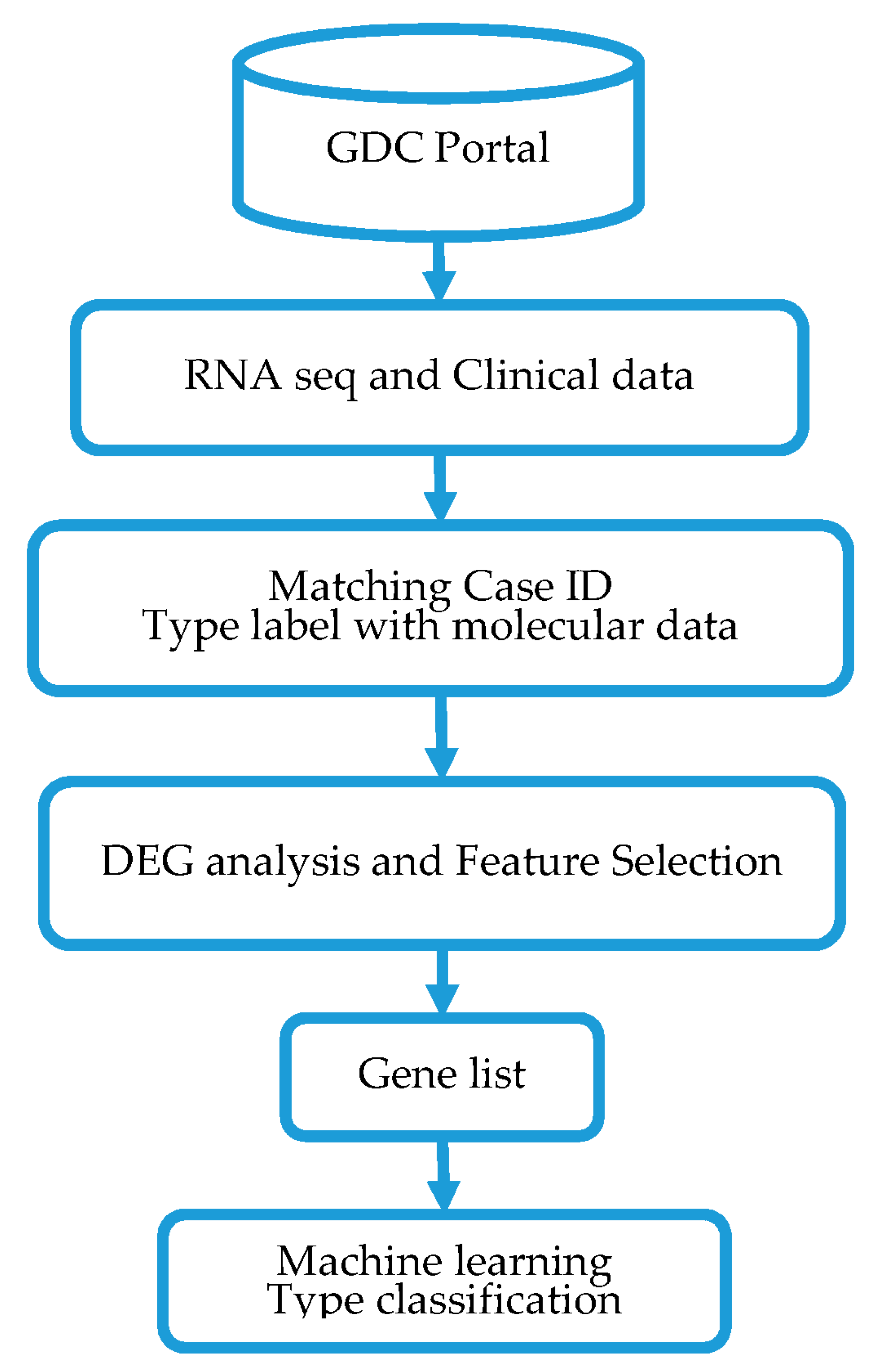
2. Materials and Methods
2.1. Source of Gene Expression Data
2.2. Differential Gene Expression Analysis and Feature Selection
2.3. Modeling Prediction and Performance Evaluation
Recall = TP/(TP + FN)
Specificity = TN/(TN + FP)
Precision = TP/(TP + FP)
F1 Score = 2 * (Recall * Precision)/(Recall + Precision)
3. Results
3.1. Result of Differential Expression and Feature Selection
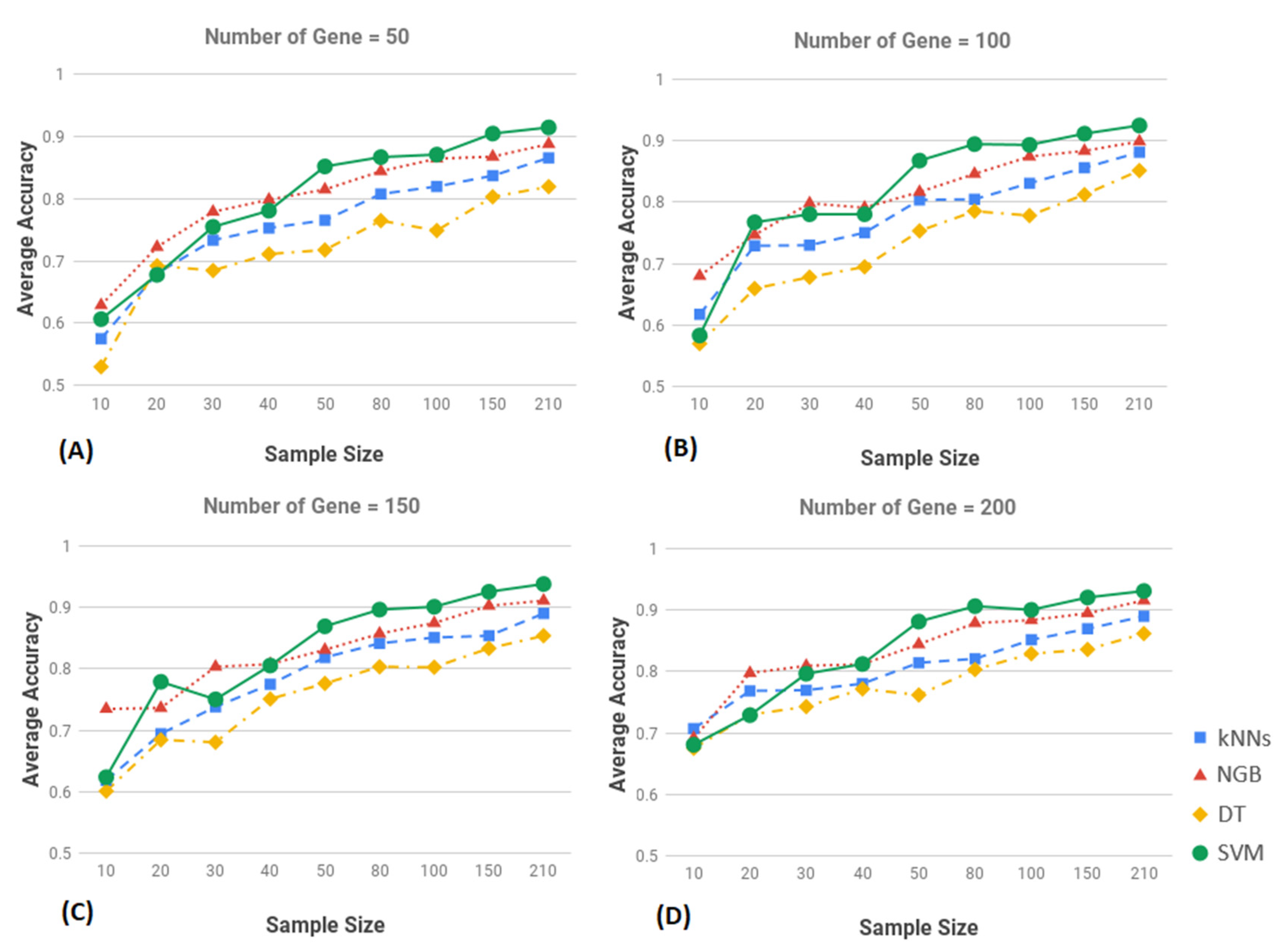
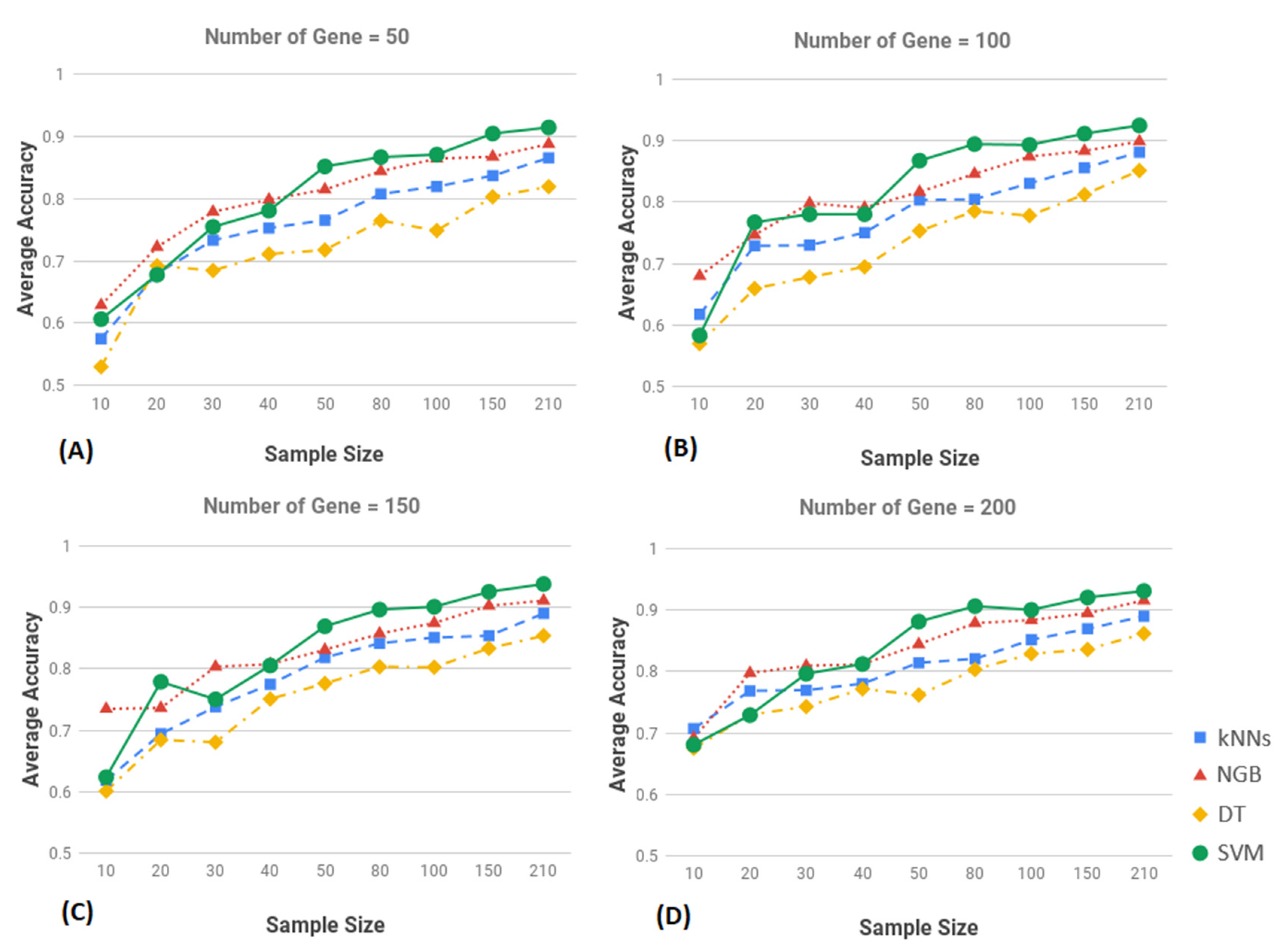
3.2. Result of Classification
3.3. Performance Evalaution of SVM
3.4. Comparative Evaluation and Validation of SVM Results
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- American Cancer Society. Cancer Facts and Figures Report 2019; American Cancer Society: Atlanta, GA, USA, 2019. [Google Scholar]
- Dietze, E.C.; Sistrunk, C.; Miranda-Carboni, G.; O’Regan, R.; Seewaldt, V.L. Triple-negative breast cancer in African-American women: Disparities versus biology. Nat. Rev. Cancer 2015, 15, 248–254. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M. Molecular Stratification of Triple-Negative Breast Cancers. Oncologist 2010, 15, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Eirew, P.; Mullaly, S.C.; Aparicio, S. The omics of triple-negative breast cancers. Clin. Chem. 2014, 60, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Homero, G., Jr.; Maximiliano, R.G.; Jane, R.; Duarte, C. Survival Study of Triple-Negative and Non-Triple-Negative Breast Cancer in a Brazilian Cohort. Clin. Med. Insights Oncol. 2018, 12, 1179554918790563. [Google Scholar]
- Joyce, D.P.; Murphy, D.; Lowery, A.J.; Curran, C.; Barry, K.; Malone, C.; McLaughlin, R.; Kerin, M.J. Prospective comparison of outcome after treatment for triple-negative and non-triple-negative breast cancer. Surgeon 2017, 15, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, J.; Peng, L.; Sahin, A.A.; Huo, L.; Ward, K.C.; O’Regan, R.; Torres, M.A.; Meisel, J.L. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 161, 279–287. [Google Scholar] [CrossRef]
- Pan, X.B.; Qu, S.; Jiang, Y.M.; Zhu, X.D. Triple Negative Breast Cancer versus Non-Triple Negative Breast Cancer Treated with Breast Conservation Surgery Followed by Radiotherapy: A Systematic Review and Meta-Analysis. Breast Care 2015, 10, 413–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Xia, X.; Dong, W.; Hao, H.; Meng, L.; Yang, Y.; Wang, R.; Lyu, Y.; Liu, Y. Cellular uptake mechanism and comparative evaluation of antineoplastic e_ects of paclitaxel-cholesterol lipid emulsion on triple-negative and non-triple-negative breast cancer cell lines. Int. J. Nanomed. 2016, 11, 4125–4140. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Xue, X.; Hu, C.; Xu, H.; Kou, D.; Li, R.; Li, M. Comparison of Clinicopathological Features and Prognosis in Triple-Negative and Non-Triple Negative Breast Cancer. J. Cancer 2016, 7, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Podo, F.; Santoro, F.; di Leo, G.; Manoukian, S.; de Giacomi, C.; Corcione, S.; Cortesi, L.; Carbonaro, L.A.; Trimboli, R.M.; Cilotti, A.; et al. Triple-Negative versus Non-Triple-Negative Breast Cancers in High-Risk Women: Phenotype Features and Survival from the HIBCRIT-1 MRI-Including Screening Study. Clin. Cancer Res. 2016, 22, 895–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabi, M.G.; Ahangar, A.; Wahid, M.A.; Kuchay, S. Clinicopathological comparison of triple negative breast cancers with non-triple negative breast cancers in a hospital in North India. Niger. J. Clin. Pract. 2015, 18, 381–386. [Google Scholar]
- Koshy, N.; Quispe, D.; Shi, R.; Mansour, R.; Burton, G.V. Cisplatin-gemcitabine therapy in metastatic breast cancer: Improved outcome in triple negative breast cancer patients compared to non-triple negative patients. Breast 2010, 19, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Milica, N.; Ana, D. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar]
- Giuseppe, V.; Leen, S.; de Snoo, F.A. Discordant assessment of tumor biomarkers by histopathological and molecular assays in the EORTC randomized controlled 10041/BIG 03-04 MINDACT trial breast cancer: Intratumoral heterogeneity and DCIS or normal tissue components are unlikely to be the cause of discordance. Breast Cancer Res. Treat. 2016, 155, 463–469. [Google Scholar]
- Viale, G.; de Snoo, F.A.; Slaets, L.; Bogaerts, J. Immunohistochemical versus molecular (BluePrint and MammaPrint) subtyping of breast carcinoma. Outcome results from the EORTC 10041/BIG 3-04 MINDACT trial. Breast Cancer Res. Treat. 2018, 167, 123–131. [Google Scholar] [CrossRef]
- Michael, U.; Bernd, G.; Nadia, H. Gallen international breast cancer conference 2013: Primary therapy of early breast cancer evidence, controversies, consensus—Opinion of a german team of experts (zurich 2013). Breast Care 2013, 8, 221–229. [Google Scholar]
- Annarita, F.; Teresa, M.B.; Liliana, L. Ensemble Discrete Wavelet Transform and Gray-Level Co-Occurrence Matrix for Microcalcification Cluster Classification in Digital Mammography. Appl. Sci. 2019, 9, 5388. [Google Scholar]
- Liliana, L.; Annarita, F.; Teresa, M.; Basile, A. Radiomics Analysis on Contrast-Enhanced Spectral Mammography Images for Breast Cancer Diagnosis: A Pilot Study. Entropy 2019, 21, 1110. [Google Scholar]
- Allegra, C.; Andrea, D.; Iole, I. Radiomics in breast cancer classification and prediction. In Seminars Cancer Biology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Mitra, M.; Mohadeseh, M.; Mahdieh, M.; Amin, B. Machine learning models in breast cancer survival prediction. Technol. Health Care 2016, 24, 31–42. [Google Scholar]
- Tong, W.; Laith, R.S.; Jiawei, T.; Theodore, W.C.; Chandra, M.S. Machine learning for diagnostic ultrasound of triple-negative breast cancer. Breast Cancer Res. Treat. 2019, 173, 365–373. [Google Scholar]
- Riku, T.; Dmitrii, B.; Mikael, L. Breast cancer outcome prediction with tumour tissue images and machine learning. Breast Cancer Res. Treat 2019, 177, 41–52. [Google Scholar]
- Weinstein, J.N.; The Cancer Genome Atlas Research Network; Collisson, E.A. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. The Genomics Data Commons. Available online: https://gdc.cancer.gov/ (accessed on 19 December 2020).
- Ritchie, M.E.; Phipson, B.; Wu, D. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Kas, K.; Schoenmakers, E.F.; Van de Ven, W.J. Physical map location of the human carboxypeptidase M gene (CPM) distal to D12S375 and proximal to D12S8 at chromosome 12q15. Genomics 1995, 30, 403–405. [Google Scholar]
- Mihaly, V.; Peter, T. The Protein Ensemble Database. Adv. Exp. Med. Biol. 2015, 870, 335–349. [Google Scholar]
- Benjamini, Y.; Yosef, H. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Shawe-Taylor, J.; Nello, C. Kernel Methods for Pattern Analysis; Cambridge University Press: Cambridge, UK, 2004; ISBN 0-521-81397-2. [Google Scholar]
- Bernhard, S.; Smola, A.J. Learning with Kernels; MIT Press: Cambridge, MA, USA, 2002; ISBN 0-262-19475-9. [Google Scholar]
- Powers, D.M.W. Evaluation: From Precision, Recall and F-Measure to ROC, Informedness, Markedness & Correlation. J. Mach. Learn. Technol. 2011, 2, 37–63. [Google Scholar]
- Huang, M.L.; Hung, Y.H.; Lee, W.M.; Li, R.K.; Jiang, B.R. SVM-RFE based feature selection and Taguchi parameters optimization for multiclass SVM classifier. Sci. World J. 2014, 795624. [Google Scholar] [CrossRef]
- Piñero, P.; Arco, L.; García, M.M.; Caballero, Y.; Yzquierdo, R.; Morales, A. Two New Metrics for Feature Selection in Pattern Recognition. In Progress in Pattern Recognition, Speech and Image Analysis. CIARP 2003. Lecture Notes in Computer Science; Sanfeliu, A., Ruiz-Shulcloper, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Kira, K.; Rendell, L. The Feature Selection Problem: Traditional Methods and a New Algorithm. In Proceedings of the AAAI-92 Proceedings, San Jose, CA, USA, 12–16 July 1992. [Google Scholar]
- Auffarth, B.; Lopez, M.; Cerquides, J. Comparison of redundancy and relevance measures for feature selection in tissue classification of CT images. In Proceedings of the Industrial Conference on Data Mining, Berlin, Germany, 12–14 July 2010; pp. 248–262. [Google Scholar]
- Tony, C.S.; Eibe, F. Introducing Machine Learning Concepts with WEKA. Methods Mol. Biol. 2016, 1418, 353–378. [Google Scholar]
- Ricvan, D.N.; Teguh, A.; Lutfan, L.; Iwan, D. Diagnostic Accuracy of Different Machine Learning Algorithms for Breast Cancer Risk Calculation: A Meta-Analysis. Asian Pac. J. Cancer Prev. 2018, 19, 1747–1752. [Google Scholar]
- La Forgia, D. Radiomic Analysis in Contrast-Enhanced Spectral Mammography for Predicting Breast Cancer Histological Outcome. Diagnostics 2020, 10, 708. [Google Scholar] [CrossRef] [PubMed]
- Asri, H.; Mousannif, H.; Al Moatassime, H.; Noel, T. Using machine learning algorithms for breast cancer risk prediction and diagnosis. Procedia Comput. Sci. 2016, 83, 1064–1069. [Google Scholar] [CrossRef] [Green Version]
- Polat, K.; Gunes, S. Breast cancer diagnosis using least square support vector machine. Digit. Signal Process 2007, 17, 694–701. [Google Scholar] [CrossRef]
- Akay, M.F. Support vector machines combined with feature selection for breast cancer diagnosis. Expert Syst. Appl. 2006, 36, 3240–3247. [Google Scholar] [CrossRef]
- Heidari, M.; Khuzani, A.Z.; Hollingsworth, A.B. Prediction of breast cancer risk using a machine learning approach embedded with a locality preserving projection algorithm. Phys. Med. Biol. 2018, 63, 035020. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Gene Name | Chromosome | Log2 Fold Change (logFC) | Adjust p-Value |
|---|---|---|---|
| ESR1 | 6q25.1-q25.2 | −8.966061547 | 1.02 × 10−35 |
| MLPH | 2q37.3 | −6.231155611 | 1.02 × 10−35 |
| FSIP1 | 15q14 | −6.785688629 | 2.04 × 10−35 |
| C5AR2 | 19q13.32 | −4.919151624 | 3.08 × 10−35 |
| GATA3 | 10p14 | −5.490221514 | 4.68 × 10−35 |
| TBC1D9 | 4q31.21 | −4.720190121 | 8.82 × 10−35 |
| CT62 | 15q23 | −8.112412605 | 9.86 × 10−35 |
| TFF1 | 21q22.3 | −13.06903719 | 2.16 × 10−34 |
| PRR15 | 7p14.3 | −6.25260355 | 2.16 × 10−34 |
| CA12 | 15q22.2 | −6.168504259 | 2.16 × 10−34 |
| AGR3 | 7p21.1 | −11.46873847 | 2.38 × 10−34 |
| SRARP | 1p36.13 | −12.26807072 | 7.31 × 10−34 |
| AGR2 | 7p21.1 | −8.8234708 | 1.32 × 10−33 |
| BCAS1 | 20q13.2 | −6.465140066 | 1.34 × 10−33 |
| LINC00504 | 4p15.33 | −7.846987181 | 2.13 × 10−33 |
| THSD4 | 15q23 | −5.0752667 | 2.13 × 10−33 |
| CCDC170 | 6q25.1 | −5.019657927 | 2.13 × 10−33 |
| RHOB | 2p24.1 | −2.828470443 | 2.13 × 10−33 |
| FOXA1 | 14q21.1 | −8.268856317 | 2.78 × 10−33 |
| ZNF552 | 19q13.43 | −3.813954916 | 2.78 × 10−33 |
| SLC16A6 | 17q24.2 | −4.45954505 | 2.99 × 10−33 |
| CFAP61 | 20p11.23 | −3.680660547 | 4.88 × 10−33 |
| GTF2IP7 | 7q11.23 | −6.49829058 | 4.98 × 10−33 |
| NEK5 | 13q14.3 | −3.666310207 | 5.90 × 10−33 |
| TTC6 | 14q21.1 | −7.69269993 | 1.00 × 10−32 |
| HID1 | 17q25.1 | −3.069655358 | 1.00 × 10−32 |
| ANXA9 | 1q21.3 | −3.748683928 | 1.45 × 10−32 |
| AK8 | 9q34.13 | −3.134793023 | 1.45 × 10−32 |
| FAM198B-AS1 | 4q32.1 | −4.757293943 | 1.63 × 10−32 |
| NAT1 | 8p22 | −6.278947772 | 3.24 × 10−32 |
| Accuracy | Recall | Specificity | |
|---|---|---|---|
| K-nearest neighbor (kNN) | 87% | 76% | 88% |
| Naïve Bayes(NGB) | 85% | 68% | 87% |
| Decision trees (DT) | 87% | 54% | 91% |
| Support Vector Machines (SVM) | 90% | 87% | 90% |
| Number of Genes | Training Set | Test Set | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy | Precision | Recall | Specify | F1 Score | Accuracy | Precision | Recall | Specify | F1 Score | |
| All (5502) | 0.90 | 0.51 | 0.87 | 0.90 | 0.65 | 0.82 | 0.33 | 0.67 | 0.80 | 0.44 |
| 4096 | 0.90 | 0.52 | 0.88 | 0.91 | 0.65 | 0.85 | 0.37 | 0.58 | 0.71 | 0.45 |
| 2048 | 0.92 | 0.56 | 0.86 | 0.92 | 0.68 | 0.84 | 0.38 | 0.75 | 0.83 | 0.50 |
| 1024 | 0.91 | 0.53 | 0.87 | 0.91 | 0.66 | 0.86 | 0.41 | 0.75 | 0.81 | 0.53 |
| 512 | 0.90 | 0.51 | 0.88 | 0.90 | 0.65 | 0.83 | 0.33 | 0.58 | 0.74 | 0.42 |
| 256 | 0.91 | 0.53 | 0.89 | 0.91 | 0.67 | 0.85 | 0.38 | 0.67 | 0.76 | 0.48 |
| 128 | 0.89 | 0.49 | 0.87 | 0.90 | 0.63 | 0.82 | 0.35 | 0.75 | 0.85 | 0.47 |
| 64 | 0.87 | 0.44 | 0.78 | 0.88 | 0.56 | 0.76 | 0.26 | 0.67 | 0.85 | 0.37 |
| 32 | 0.78 | 0.27 | 0.64 | 0.80 | 0.38 | 0.71 | 0.19 | 0.50 | 0.81 | 0.27 |
| 16 | 0.74 | 0.22 | 0.63 | 0.75 | 0.33 | 0.69 | 0.21 | 0.67 | 0.89 | 0.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Hicks, C. Breast Cancer Type Classification Using Machine Learning. J. Pers. Med. 2021, 11, 61. https://doi.org/10.3390/jpm11020061
Wu J, Hicks C. Breast Cancer Type Classification Using Machine Learning. Journal of Personalized Medicine. 2021; 11(2):61. https://doi.org/10.3390/jpm11020061
Chicago/Turabian StyleWu, Jiande, and Chindo Hicks. 2021. "Breast Cancer Type Classification Using Machine Learning" Journal of Personalized Medicine 11, no. 2: 61. https://doi.org/10.3390/jpm11020061
APA StyleWu, J., & Hicks, C. (2021). Breast Cancer Type Classification Using Machine Learning. Journal of Personalized Medicine, 11(2), 61. https://doi.org/10.3390/jpm11020061