
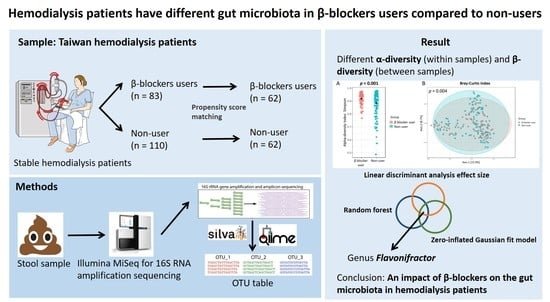
Differences in the Microbial Composition of Hemodialysis Patients Treated with and without ?-Blockers

 ,
,  , , , , ,
, , , , , 
Abstract
:
1. Introduction
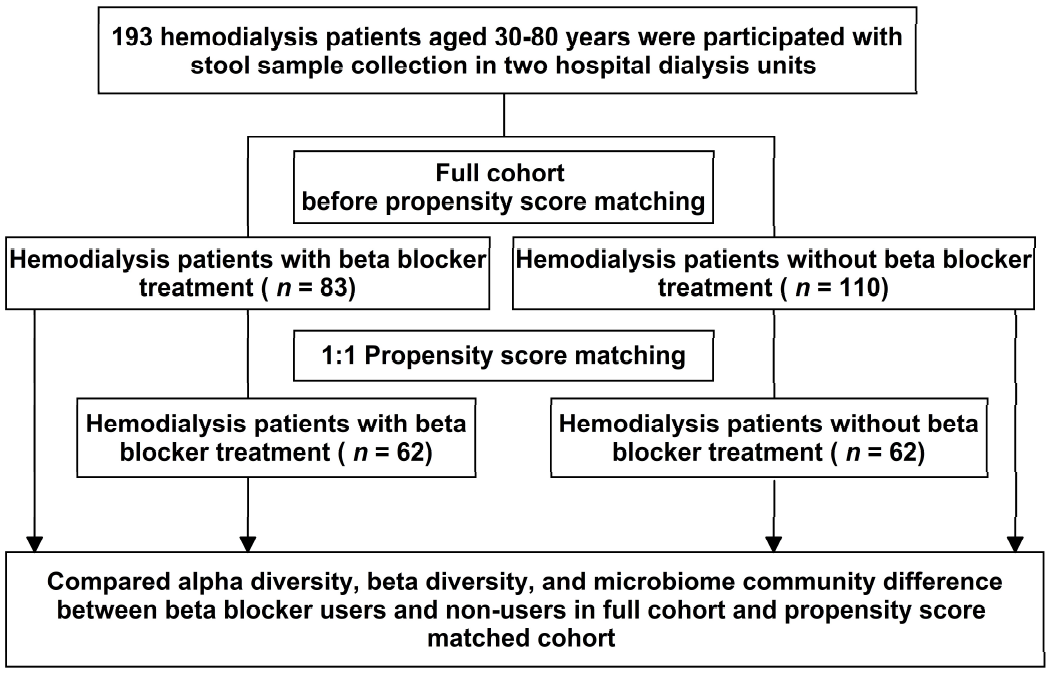
2. Materials and Methods
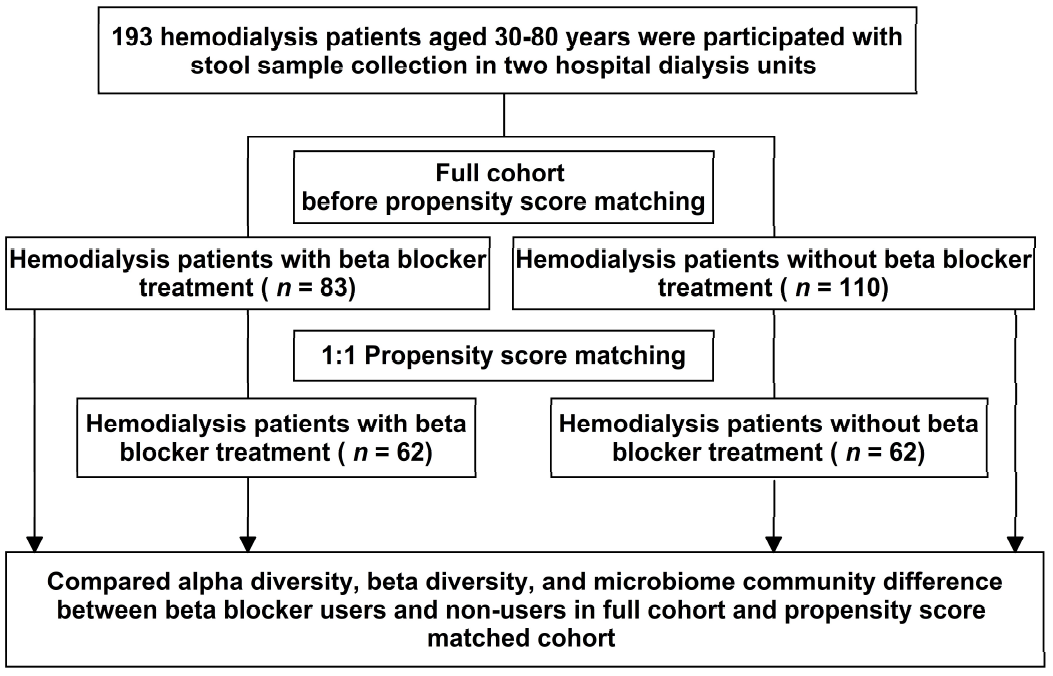
2.1. Study Participants
2.2. Comorbidity, Laboratory, and Clinical Variables
2.3. Fecal Sample Collection and Bacterial 16S rRNA Amplicon Sequencing and Processing
2.4. Propensity Score Matching
2.5. Statistical and Bioinformatics Analyses of Microbiota
2.6. Functional Prediction Analysis
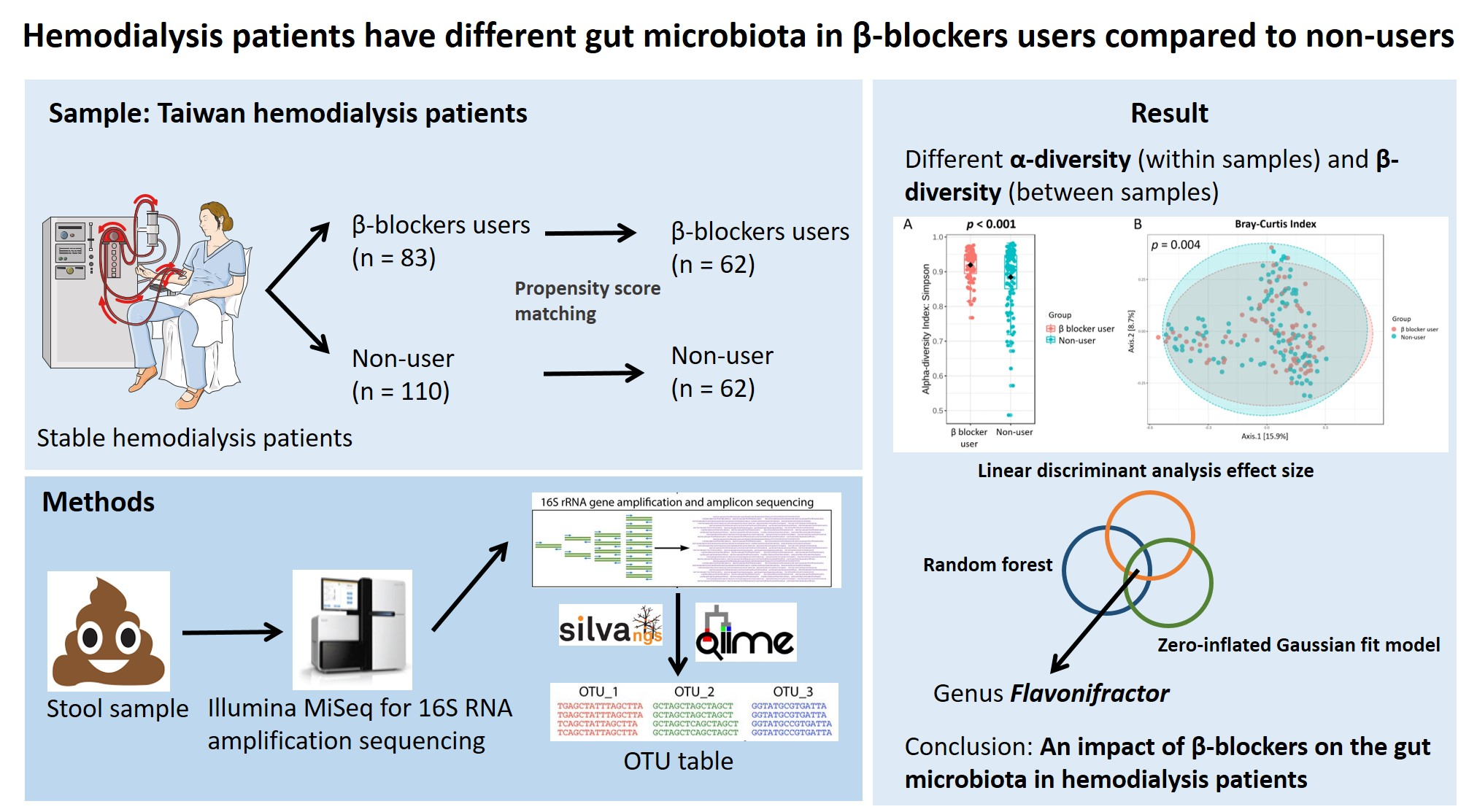
3. Results
3.1. Patient Characteristics
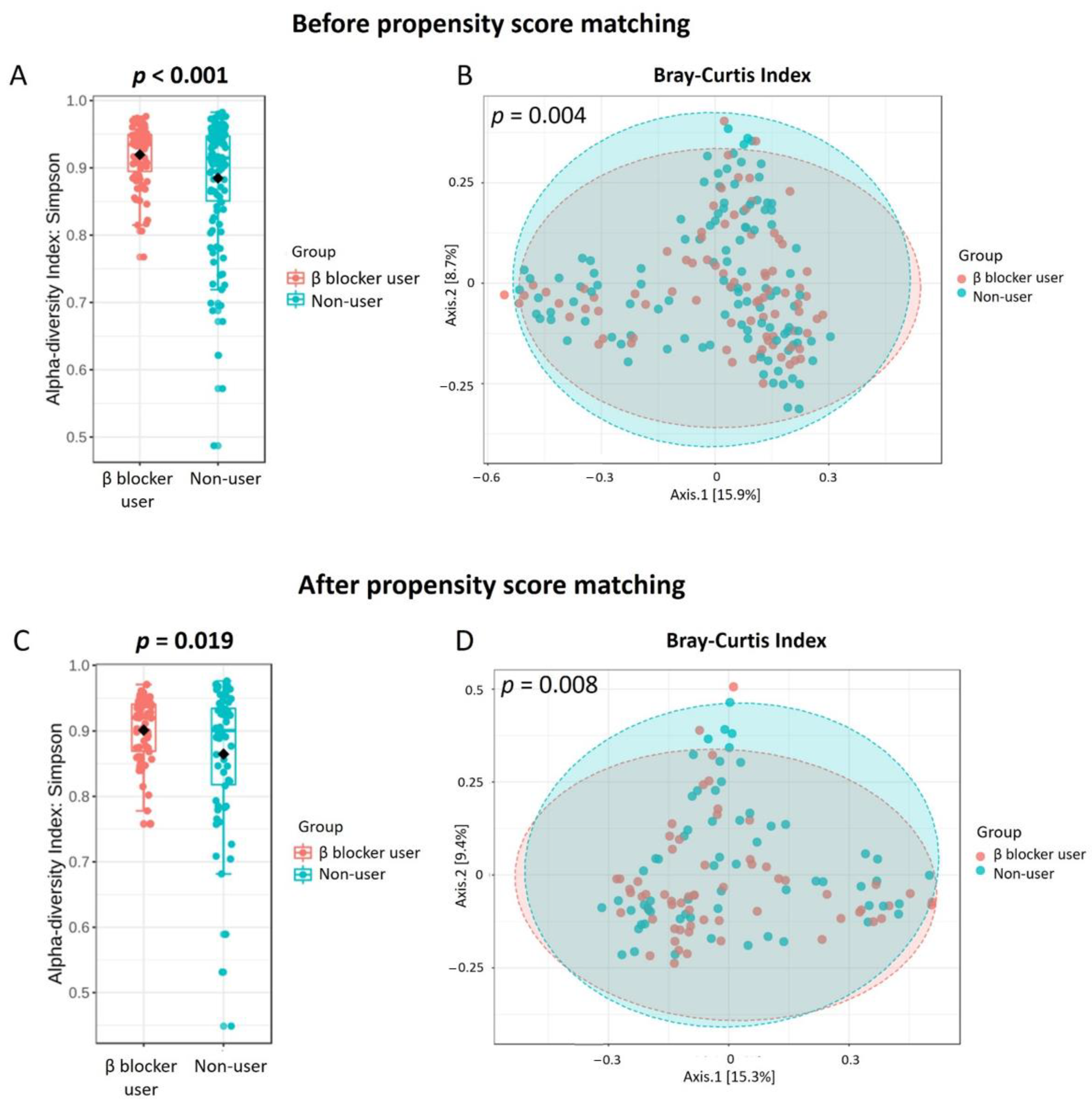
3.2. Gut Microbiota Profile Differs in Hemodialysis Patients with and without β Blocker Treatment
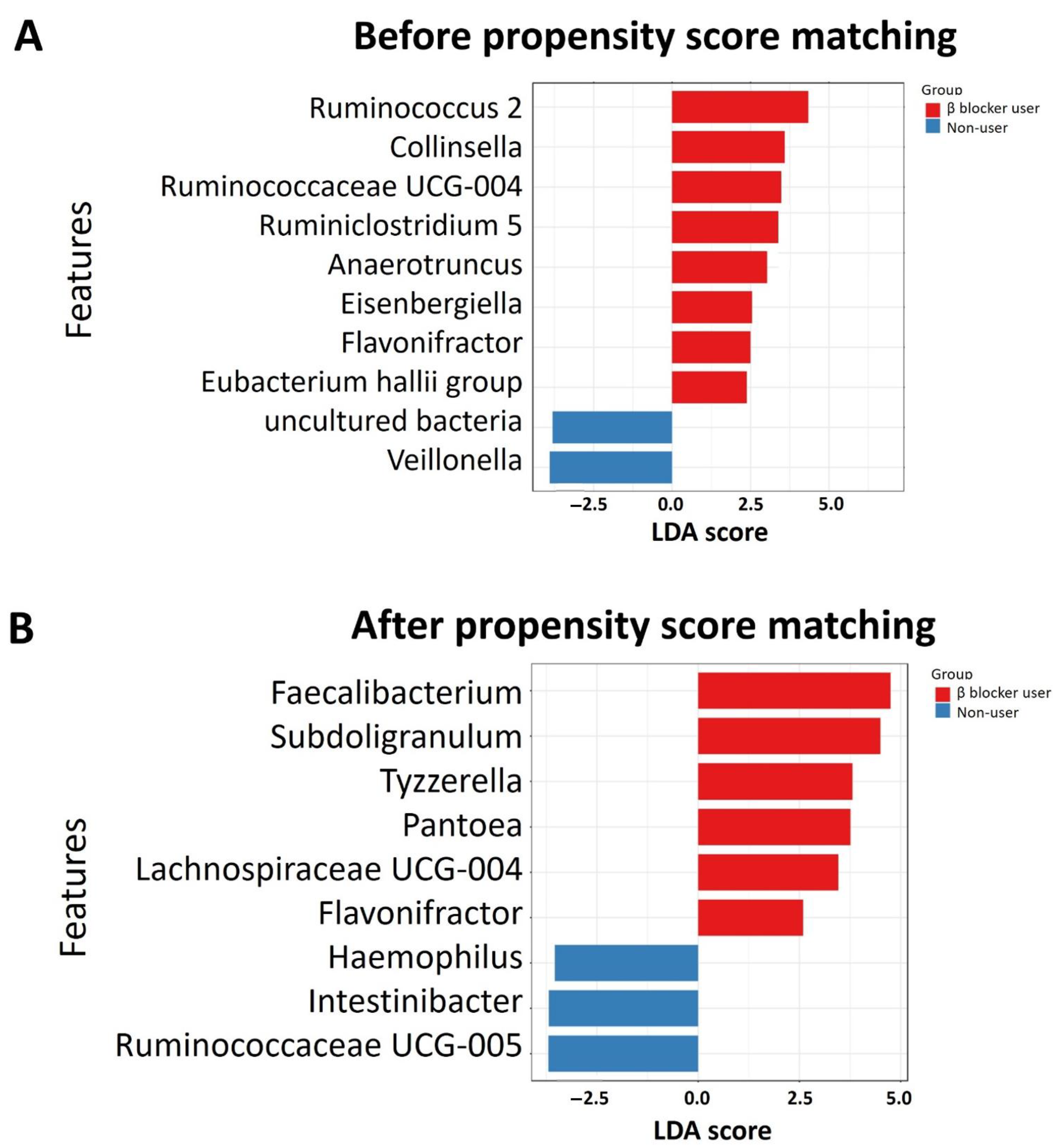
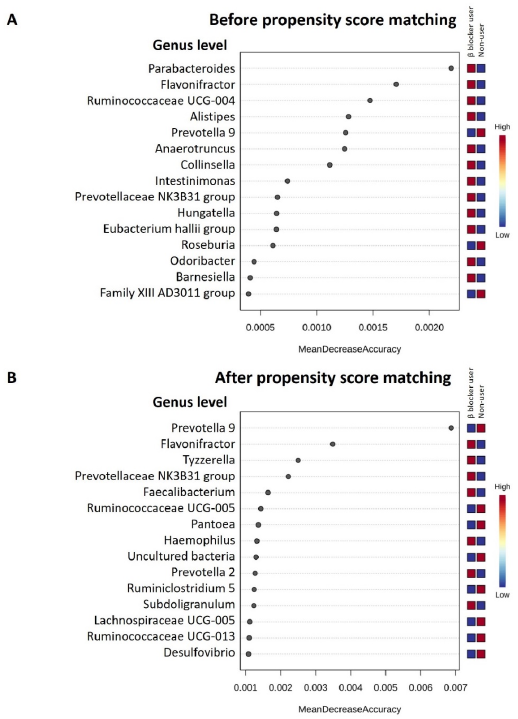
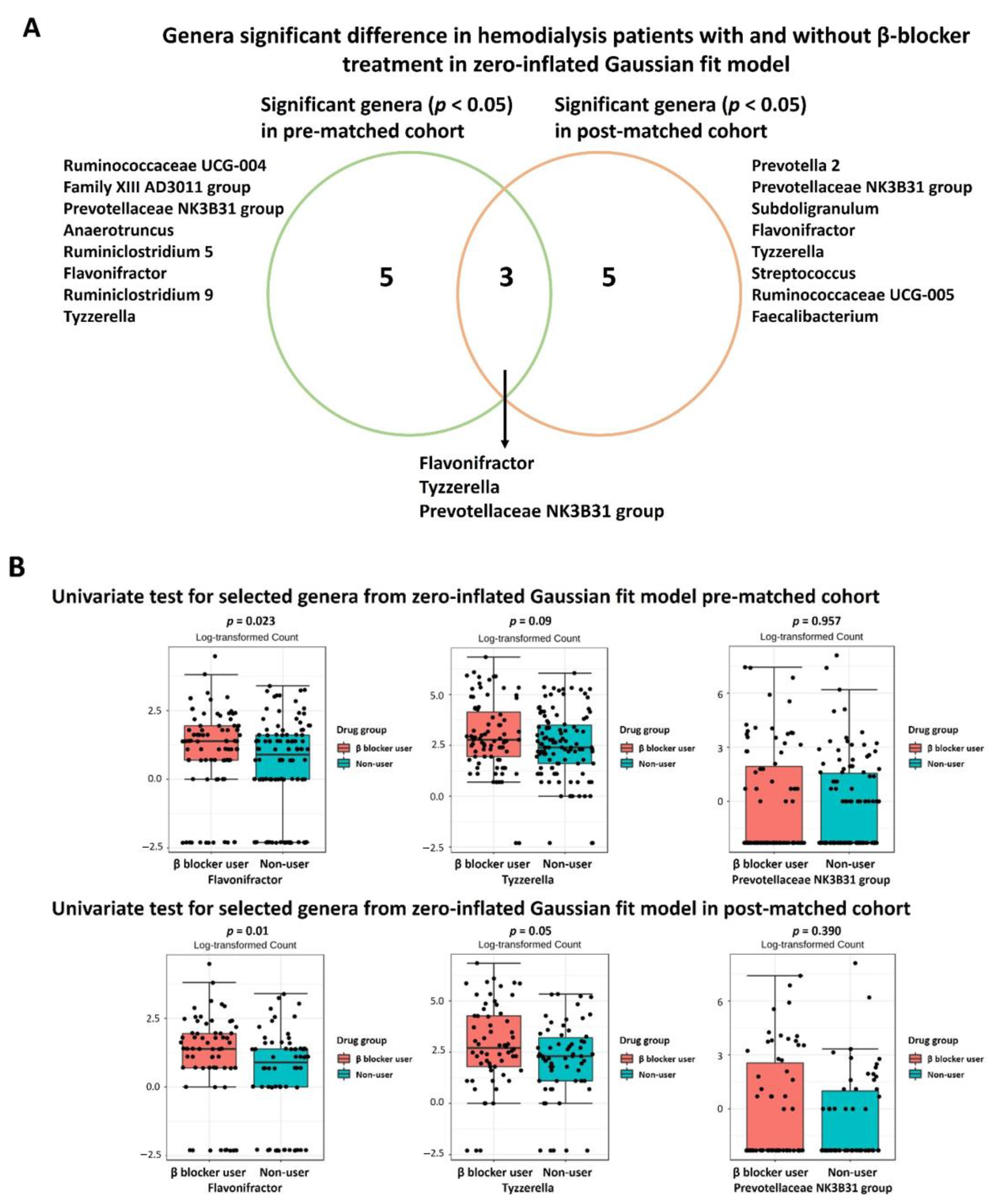
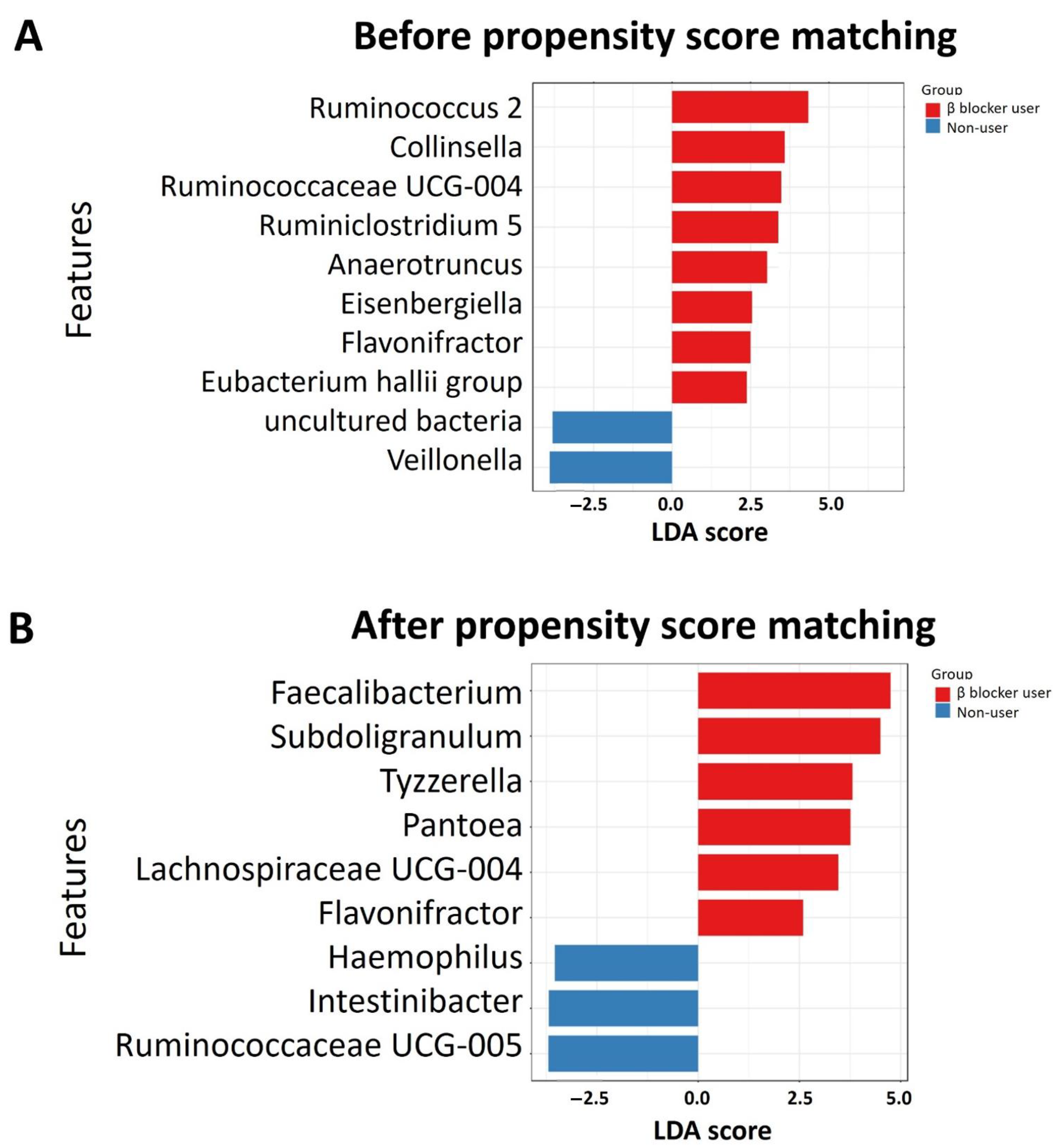
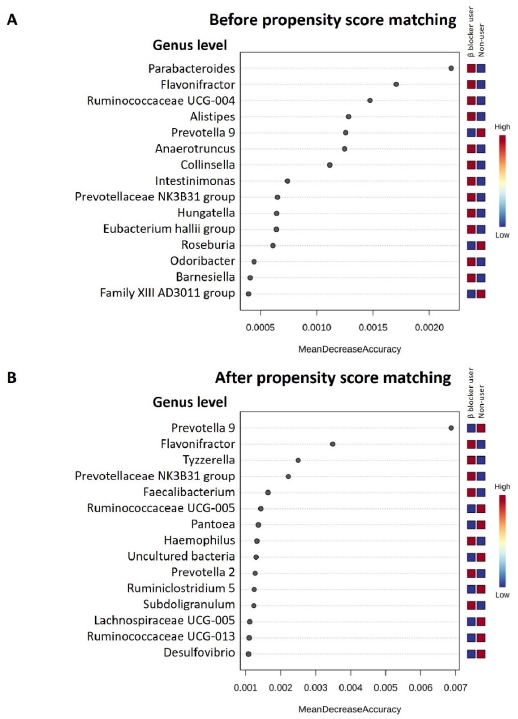
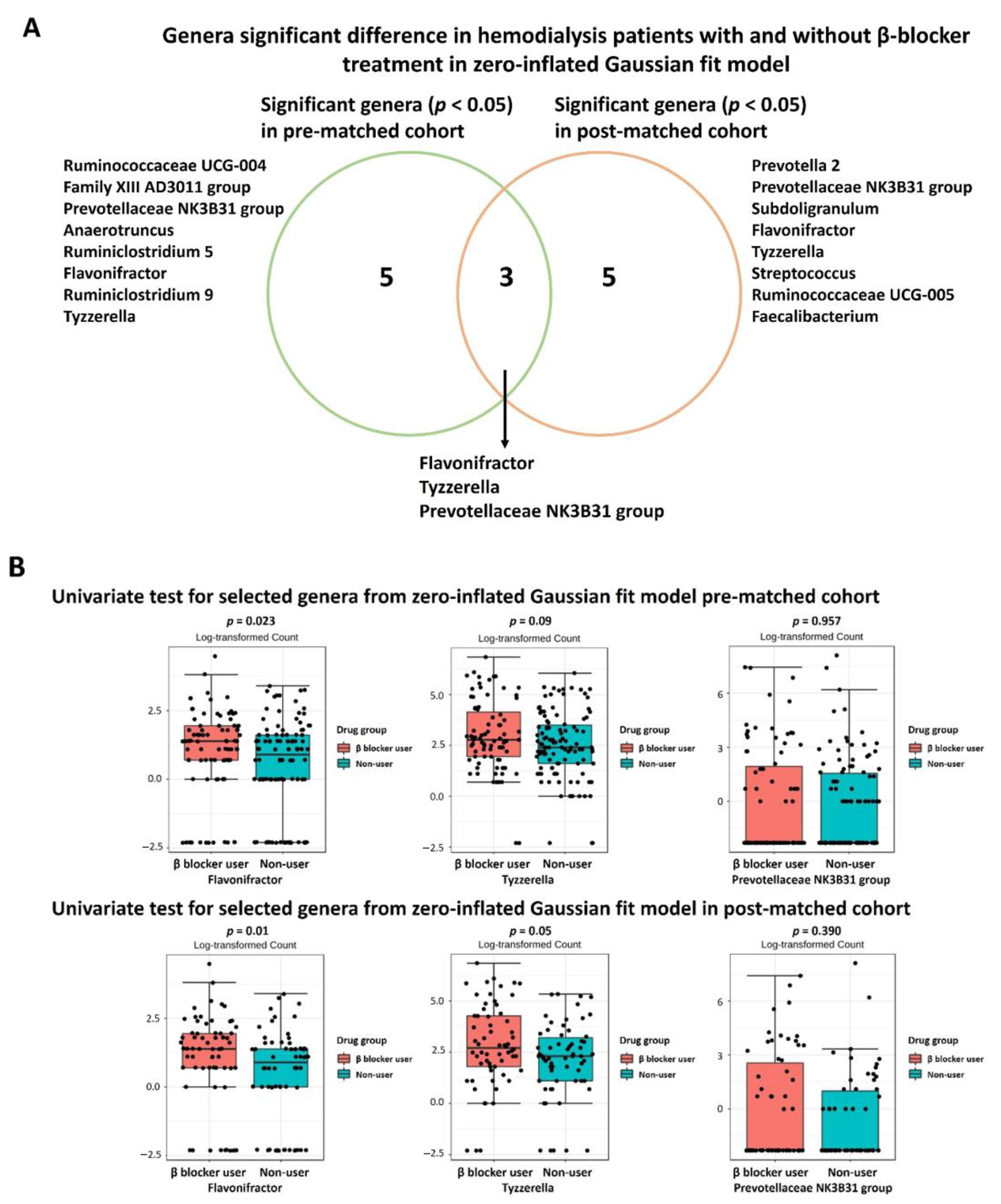
3.3. Specific Microbial Taxa Differences between β-Blocker Users and Nonusers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Salvucci, E. The human-microbiome superorganism and its modulation to restore health. Int. J. Food Sci. Nutr. 2019, 70, 781–795. [Google Scholar] [CrossRef]
- Belizario, J.E.; Faintuch, J.; Garay-Malpartida, M. Gut Microbiome Dysbiosis and Immunometabolism: New Frontiers for Treatment of Metabolic Diseases. Mediators Inflamm. 2018, 2018, 2037838. [Google Scholar] [CrossRef] [PubMed]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel. Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [Green Version]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef]
- Jackson, M.A.; Verdi, S.; Maxan, M.E.; Shin, C.M.; Zierer, J.; Bowyer, R.C.E.; Martin, T.; Williams, F.M.K.; Menni, C.; Bell, J.T.; et al. Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nat. Commun. 2018, 9, 2655. [Google Scholar] [CrossRef] [Green Version]
- Imhann, F.; Bonder, M.J.; Vich Vila, A.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.A.; Cenit, M.C.; Harmsen, H.J.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makivuokko, H.; Tiihonen, K.; Tynkkynen, S.; Paulin, L.; Rautonen, N. The effect of age and non-steroidal anti-inflammatory drugs on human intestinal microbiota composition. Br. J. Nutr. 2010, 103, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahr, S.M.; Tyler, B.C.; Wooldridge, N.; Butcher, B.D.; Burns, T.L.; Teesch, L.M.; Oltman, C.L.; Azcarate-Peril, M.A.; Kirby, J.R.; Calarge, C.A. Use of the second-generation antipsychotic, risperidone, and secondary weight gain are associated with an altered gut microbiota in children. Transl. Psychiatry 2015, 5, e652. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Freedberg, D.E.; Toussaint, N.C.; Chen, S.P.; Ratner, A.J.; Whittier, S.; Wang, T.C.; Wang, H.H.; Abrams, J.A. Proton Pump Inhibitors Alter Specific Taxa in the Human Gastrointestinal Microbiome: A Crossover Trial. Gastroenterology 2015, 149, 883–885.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, M.A.; Goodrich, J.K.; Maxan, M.E.; Freedberg, D.E.; Abrams, J.A.; Poole, A.C.; Sutter, J.L.; Welter, D.; Ley, R.E.; Bell, J.T.; et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut 2016, 65, 749–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Pamer, E.G. Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol. 2013, 13, 790–801. [Google Scholar] [CrossRef] [Green Version]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Poesen, R.; Meijers, B. The gut-kidney axis. Pediatr. Nephrol. 2017, 32, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Weir, M.A.; Herzog, C.A. Beta blockers in patients with end-stage renal disease-Evidence-based recommendations. Semin. Dial. 2018, 31, 219–225. [Google Scholar] [CrossRef]
- Bakris, G.L.; Hart, P.; Ritz, E. Beta blockers in the management of chronic kidney disease. Kidney Int. 2006, 70, 1905–1913. [Google Scholar] [CrossRef] [Green Version]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.e22. [Google Scholar] [CrossRef]
- Ikizler, T.A.; Burrowes, J.D.; Byham-Gray, L.D.; Campbell, K.L.; Carrero, J.J.; Chan, W.; Fouque, D.; Friedman, A.N.; Ghaddar, S.; Goldstein-Fuchs, D.J.; et al. KDOQI Clinical Practice Guideline for Nutrition in CKD: 2020 Update. Am. J. Kidney Dis. 2020, 76, S1–S107. [Google Scholar] [CrossRef] [PubMed]
- Herlemann, D.P.; Labrenz, M.; Jurgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G. Microbiome Helper: A Custom and Streamlined Workflow for Microbiome Research. mSystems 2017, 2, e00127-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic. Acids. Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glockner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic. Acids. Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Sturmer, T.; Wyss, R.; Glynn, R.J.; Brookhart, M.A. Propensity scores for confounder adjustment when assessing the effects of medical interventions using nonexperimental study designs. J. Intern. Med. 2014, 275, 570–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.S.; Groenwold, R.H.; Belitser, S.V.; Pestman, W.R.; Hoes, A.W.; Roes, K.C.; Boer, A.; Klungel, O.H. Reporting of covariate selection and balance assessment in propensity score analysis is suboptimal: A systematic review. J. Clin. Epidemiol. 2015, 68, 112–121. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [Green Version]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Markowitz, V.M.; Chen, I.M.; Palaniappan, K.; Chu, K.; Szeto, E.; Grechkin, Y.; Ratner, A.; Jacob, B.; Huang, J.; Williams, P.; et al. IMG: The Integrated Microbial Genomes database and comparative analysis system. Nucleic. Acids. Res. 2012, 40, D115–D122. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.-Y. poyuliu/MARco: MARco: Microbiome Analysis RcodeDB (Version v1.0). Zenodo. Available online: http://doi.org/10.5281/zenodo.4589898 (accessed on 1 February 2021).
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [Green Version]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.L.; Vaziri, N.D.; Nunes, A.C.F.; Comeau, A.M.; Langille, M.G.I.; England, W.; Khazaeli, M.; Suematsu, Y.; Phan, J.; Whiteson, K. The Phosphate Binder Ferric Citrate Alters the Gut Microbiome in Rats with Chronic Kidney Disease. J. Pharmacol. Exp. Ther. 2018, 367, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Lun, H.; Yang, W.; Zhao, S.; Jiang, M.; Xu, M.; Liu, F.; Wang, Y. Altered gut microbiota and microbial biomarkers associated with chronic kidney disease. Microbiologyopen 2019, 8, e00678. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American Gut: An Open Platform for Citizen Science Microbiome Research. mSystems 2018, 3, e00031-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Roy, C.I.; Wells, P.M.; Si, J.; Raes, J.; Bell, J.T.; Spector, T.D. Red Wine Consumption Associated With Increased Gut Microbiota alpha-Diversity in 3 Independent Cohorts. Gastroenterology 2020, 158, 270–272. [Google Scholar] [CrossRef]
- Leeming, E.R.; Johnson, A.J.; Spector, T.D.; Le Roy, C.I. Effect of Diet on the Gut Microbiota: Rethinking Intervention Duration. Nutrients 2019, 11, 2862. [Google Scholar] [CrossRef] [Green Version]
- Le Bastard, Q.; Al-Ghalith, G.A.; Gregoire, M.; Chapelet, G.; Javaudin, F.; Dailly, E.; Batard, E.; Knights, D.; Montassier, E. Systematic review: Human gut dysbiosis induced by non-antibiotic prescription medications. Aliment. Pharmacol. Ther. 2018, 47, 332–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y.; et al. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015, 15, 100. [Google Scholar] [CrossRef] [Green Version]
- Zuo, K.; Li, J.; Li, K.; Hu, C.; Gao, Y.; Chen, M.; Hu, R.; Liu, Y.; Chi, H.; Wang, H.; et al. Disordered gut microbiota and alterations in metabolic patterns are associated with atrial fibrillation. Gigascience 2019, 8, giz058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Gao, R.; Zhang, Y.; Pan, D.; Zhu, Y.; Zhang, X.; Yang, R.; Jiang, R.; Xu, Y.; Qin, H. Dysbiosis signatures of gut microbiota in coronary artery disease. Physiol. Genom. 2018, 50, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef] [Green Version]
- Carlier, J.P.; Bedora-Faure, M.; K’Ouas, G.; Alauzet, C.; Mory, F. Proposal to unify Clostridium orbiscindens Winter et al. 1991 and Eubacterium plautii (Seguin 1928) Hofstad and Aasjord 1982, with description of Flavonifractor plautii gen. nov., comb. nov., and reassignment of Bacteroides capillosus to Pseudoflavonifractor capillosus gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2010, 60, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Coello, K.; Hansen, T.H.; Sorensen, N.; Munkholm, K.; Kessing, L.V.; Pedersen, O.; Vinberg, M. Gut microbiota composition in patients with newly diagnosed bipolar disorder and their unaffected first-degree relatives. Brain Behav. Immun. 2019, 75, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Mao, J.; Zhou, L.; Xiong, X.; Deng, Y. The imbalance of gut microbiota and its correlation with plasma inflammatory cytokines in pemphigus vulgaris patients. Scand. J. Immunol. 2019, 90, e12799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikami, A.; Ogita, T.; Namai, F.; Shigemori, S.; Sato, T.; Shimosato, T. Oral administration of Flavonifractor plautii attenuates inflammatory responses in obese adipose tissue. Mol. Biol. Rep. 2020, 47, 6717–6725. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.N.; Bazzano, L.A.; Ajami, N.J.; He, H.; Zhao, J.; Petrosino, J.F.; Correa, A.; He, J. Gut Microbiome Associates With Lifetime Cardiovascular Disease Risk Profile Among Bogalusa Heart Study Participants. Circ. Res. 2016, 119, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Ahmari, N.; Schmidt, J.T.; Redler, T.; Arocha, R.; Pacholec, K.; Magee, K.L.; Malphurs, W.; Owen, J.L.; Krane, G.A.; et al. Shifts in the Gut Microbiota Composition Due to Depleted Bone Marrow Beta Adrenergic Signaling Are Associated with Suppressed Inflammatory Transcriptional Networks in the Mouse Colon. Front. Physiol. 2017, 8, 220. [Google Scholar] [CrossRef] [Green Version]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Dan, X.; Mushi, Z.; Baili, W.; Han, L.; Enqi, W.; Huanhu, Z.; Shuchun, L. Differential Analysis of Hypertension-Associated Intestinal Microbiota. Int. J. Med. Sci. 2019, 16, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Robles-Vera, I.; Toral, M.; Duarte, J. Microbiota and Hypertension: Role of the Sympathetic Nervous System and the Immune System. Am. J. Hypertens. 2020, 33, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Palmu, J.; Lahti, L.; Niiranen, T. Targeting Gut Microbiota to Treat Hypertension: A Systematic Review. Int. J. Environ. Res. Public Health 2021, 18, 1248. [Google Scholar] [CrossRef]
- Liu, C.; Shao, W.; Gao, M.; Liu, J.; Guo, Q.; Jin, J.; Meng, F. Changes in intestinal flora in patients with type 2 diabetes on a low-fat diet during 6 months of follow-up. Exp. Ther. Med. 2020, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Whang, A.; Nagpal, R.; Yadav, H. Bi-directional drug-microbiome interactions of anti-diabetics. EBioMedicine 2019, 39, 591–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, K.; Li, J.; Xu, Q.; Hu, C.; Gao, Y.; Chen, M.; Hu, R.; Liu, Y.; Chi, H.; Yin, Q.; et al. Dysbiotic gut microbes may contribute to hypertension by limiting vitamin D production. Clin. Cardiol. 2019, 42, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Durgan, D.J.; Ganesh, B.P.; Cope, J.L.; Ajami, N.J.; Phillips, S.C.; Petrosino, J.F.; Hollister, E.B.; Bryan, R.M., Jr. Role of the Gut Microbiome in Obstructive Sleep Apnea-Induced Hypertension. Hypertension 2016, 67, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbo, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [Green Version]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Bork, P. A fair comparison. Nat. Methods 2014, 11, 359. [Google Scholar] [CrossRef]
- Jonsson, V.; Osterlund, T.; Nerman, O.; Kristiansson, E. Modelling of zero-inflation improves inference of metagenomic gene count data. Stat. Methods Med. Res. 2019, 28, 3712–3728. [Google Scholar] [CrossRef]
- Xu, L.; Paterson, A.D.; Turpin, W.; Xu, W. Assessment and Selection of Competing Models for Zero-Inflated Microbiome Data. PLoS ONE 2015, 10, e0129606. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline Characteristics | Before Propensity Score Matching | After Propensity Score Matching | ||||
|---|---|---|---|---|---|---|
| β-Blocker Users (N = 83) | β-Blocker Nonusers (N = 110) | p-Value | β-Blocker Users (N = 62) | β-Blocker Nonusers (N = 62) | p-Value | |
| Age (years) | 64.3 ± 11.4 | 65.4 ± 11.2 | 0.511 | 64.7 ± 11.6 | 66.3 ± 11.8 | 0.446 |
| Male | 49 (59.0%) | 57 (51.8%) | 0.318 | 37 (59.7%) | 28 (45.2% | 0.106 |
| Body mass index | 23.4 ± 3.25 | 23.6 ± 3.91 | 0.708 | 23.5 ± 3.34 | 23.5 ± 3.93 | 0.988 |
| Dialysis vintage (months) | 86.24 ± 56.53 | 96.54 ± 63.21 | 0.243 | 93.22 ± 57.61 | 85.4 ± 55.67 | 0.444 |
| Smoking history | 15 (18.1%) | 12 (10.9%) | 0.156 | 9 (14.5%) | 6 (9.7%) | 0.409 |
| Arteriovenous fistula | 75 (90.4%) | 99 (90.0%) | 0.934 | 57 (91.9%) | 57 (91.9%) | >0.999 |
| Comorbidities | ||||||
| Diabetes mellitus | 45 (54.2%) | 34 (30.9%) | 0.001 | 24 (38.7%) | 30 (48.4%) | 0.277 |
| Hypertension | 80 (96.4%) | 87 (79.1%) | <0.001 | 59 (95.2%) | 59 (95.2%) | >0.999 |
| Dyslipidemia | 31 (37.3%) | 24 (21.8%) | 0.018 | 16 (25.8%) | 15 (24.2%) | 0.836 |
| Coronary artery disease | 34 (41.0%) | 22 (20.0%) | 0.001 | 21 (33.9%) | 18 (29.0%) | 0.562 |
| Heart failure | 22 (26.5%) | 15 (13.6%) | 0.025 | 14 (22.6%) | 11 (17.7%) | 0.502 |
| Cerebrovascular disease | 31 (37.3%) | 24 (21.8%) | 0.018 | 5 (8.1%) | 8 (12.9%) | 0.379 |
| Parathyroidectomy history | 7 (8.4%) | 18 (16.4%) | 0.104 | 6 (9.7%) | 6 (9.7%) | >0.999 |
| Medications | ||||||
| ACEI/ARB | 29 (34.9%) | 24 (21.8%) | 0.043 | 23 (37.1%) | 15 (24.2%) | 0.119 |
| Glucose lowering drugs | 34 (41.0%) | 23 (20.9%) | 0.003 | 20 (32.3%) | 19 (30.6%) | 0.847 |
| Sulfonylurea | 14 (16.9%) | 13 (11.8%) | 0.317 | 6 (9.7%) | 11 (17.7%) | 0.192 |
| Dipeptidyl peptidase 4 inhibitors | 28 (33.7%) | 13 (11.8%) | <0.001 | 17 (27.4%) | 11 (17.7%) | 0.198 |
| Insulin | 17 (20.5%) | 10 (9.1%) | 0.024 | 9 (14.5%) | 8 (12.9%) | 0.794 |
| Statin | 29 (34.9%) | 17 (15.5%) | 0.002 | 17 (27.4%) | 12 (19.4%) | 0.289 |
| Calcium carbonate | 67 (80.7%) | 94 (85.5%) | 0.382 | 51 (82.3%) | 50 (80.6%) | 0.817 |
| Proton pump inhibitors | 13 (15.7%) | 10 (9.1%) | 0.163 | 9 (14.5%) | 7 (11.3%) | 0.592 |
| Clinical laboratory data | ||||||
| Hemoglobin (g/dL) | 10.62 ± 1.14 | 10.71 ± 1.41 | 0.650 | 10.6 ± 1.05 | 10.74 ± 1.49 | 0.555 |
| Albumin (g/dL) | 3.52 ± 0.51 | 3.56 ± 0.46 | 0.538 | 3.53 ± 0.46 | 3.54 ± 0.47 | 0.902 |
| Total cholesterol (mg/dL) | 154.01 ± 33.75 | 161.89 ± 33.62 | 0.109 | 151.94 ± 33.57 | 163.51 ± 35.30 | 0.064 |
| Triglyceride (mg/dL) | 140.52 ± 103.77 | 129.61 ± 90.35 | 0.437 | 136.21 ± 105.99 | 131.14 ± 95.51 | 0.780 |
| High sensitivity CRP (mg/dL) | 2.15 ± 4.65 | 2.5 ± 4.21 | 0.589 | 2.45 ± 5.23 | 2.21 ± 3.95 | 0.779 |
| Sodium (mmol/L) | 136.92 ± 2.68 | 137.07 ± 2.62 | 0.700 | 137.19 ± 2.80 | 136.64 ± 2.44 | 0.241 |
| Potassium (mmol/L) | 4.73 ± 0.68 | 4.61 ± 0.62 | 0.195 | 4.77 ± 0.66 | 4.65 ± 0.65 | 0.294 |
| Total calcium (mg/dL) | 9.15 ± 0.86 | 9.29 ± 0.94 | 0.277 | 9.19 ± 0.92 | 9.25 ± 0.86 | 0.683 |
| Phosphate (mg/dL) | 5.08 ± 1.21 | 4.95 ± 1.24 | 0.453 | 5.16 ± 1.15 | 5.09 ± 1.35 | 0.768 |
| Parathyroid hormone (pg/mL) | 376.53 ± 338.79 | 383.5 ± 278.13 | 0.876 | 394.16 ± 370.62 | 357.29 ± 245.84 | 0.515 |
| Serum iron (μg/dL) | 63.57 ± 26.73 | 65.85 ± 21.16 | 0.508 | 63.94 ± 26.61 | 67.52 ± 22.93 | 0.424 |
| Ferritin (ng/mL) | 567.53 ± 549.64 | 496.67 ± 377.33 | 0.291 | 534.93 ± 330.67 | 538.54 ± 413.54 | 0.957 |
| nPCR (g/kg/day) | 1.12 ± 0.21 | 1.16 ± 0.27 | 0.326 | 1.12 ± 0.20 | 1.18 ± 0.28 | 0.180 |
| Single pool Kt/V | 1.67 ± 0.27 | 1.65 ± 0.27 | 0.591 | 1.67 ± 0.28 | 1.68 ± 0.27 | 0.817 |
| Dietary intake (serving/day) | ||||||
| Meat | 0.86 ± 0.57 | 0.82 ± 0.53 | 0.652 | 0.86 ± 0.57 | 0.74 ± 0.52 | 0.241 |
| Vegetable | 2.01 ± 1.09 | 1.86 ± 1.11 | 0.265 | 2.05 ± 1.06 | 1.91 ± 1.18 | 0.499 |
| Fruit | 0.93 ± 0.72 | 0.95 ± 0.72 | 0.583 | 0.86 ± 0.63 | 0.89 ± 0.75 | 0.837 |
| Bristol stool scale | 3.94 ± 1.86 | 3.74 ± 1.76 | 0.448 | 4 ± 1.78 | 3.71 ± 1.67 | 0.352 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-T.; Lin, T.-Y.; Hung, S.-C.; Liu, P.-Y.; Hung, W.-C.; Tsai, W.-C.; Tsai, Y.-C.; Delicano, R.A.; Chuang, Y.-S.; Kuo, M.-C.; et al. Differences in the Microbial Composition of Hemodialysis Patients Treated with and without ?-Blockers. J. Pers. Med. 2021, 11, 198. https://doi.org/10.3390/jpm11030198
Lin Y-T, Lin T-Y, Hung S-C, Liu P-Y, Hung W-C, Tsai W-C, Tsai Y-C, Delicano RA, Chuang Y-S, Kuo M-C, et al. Differences in the Microbial Composition of Hemodialysis Patients Treated with and without ?-Blockers. Journal of Personalized Medicine. 2021; 11(3):198. https://doi.org/10.3390/jpm11030198
Chicago/Turabian StyleLin, Yi-Ting, Ting-Yun Lin, Szu-Chun Hung, Po-Yu Liu, Wei-Chun Hung, Wei-Chung Tsai, Yi-Chun Tsai, Rachel Ann Delicano, Yun-Shiuan Chuang, Mei-Chuan Kuo, and et al. 2021. "Differences in the Microbial Composition of Hemodialysis Patients Treated with and without ?-Blockers" Journal of Personalized Medicine 11, no. 3: 198. https://doi.org/10.3390/jpm11030198
APA StyleLin, Y.-T., Lin, T.-Y., Hung, S.-C., Liu, P.-Y., Hung, W.-C., Tsai, W.-C., Tsai, Y.-C., Delicano, R. A., Chuang, Y.-S., Kuo, M.-C., Chiu, Y.-W., & Wu, P.-H. (2021). Differences in the Microbial Composition of Hemodialysis Patients Treated with and without ?-Blockers. Journal of Personalized Medicine, 11(3), 198. https://doi.org/10.3390/jpm11030198