
The Promise of Patient-Derived Preclinical Models to Accelerate the Implementation of Personalised Medicine for Children with Neuroblastoma

 , and
, and
Abstract
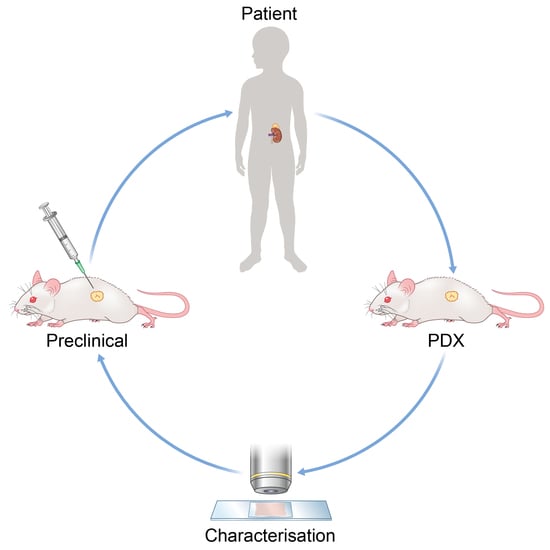
:
{kind=link}
{kind=link}
1. Introduction
2. Ethical Considerations for the Development of Patient-Derived Xenografts
3. Technical Considerations for the Development of Patient-Derived Xenografts
4. The Development and Refinement of Patient-Derived Models for Neuroblastoma
4.1. Patient-Derived Neuroblastoma Xenografts
- Subclones with very few mutations confined to a single tumour region
- Stable coexistence, over vast areas of clones characterised by chromosomal number aberrations (this was demonstrated by half of the neuroblastomas in the study)
- A clone with a driver mutation or structural rearrangement emerges through a clonal sweep to dominate an anatomical region (again, this was shown in half of neuroblastomas in the study)
- Local emergence of a myriad of clones with TP53 inactivation.
4.2. Application of Patient-Derived Neuroblastoma Xenografts
4.3. Advancing Neuroblastoma Immunotherapy Using Patient-Derived Models
4.4. Patient-Derived Neuroblastoma Zebrafish Models
4.5. Patient-Derived Neuroblastoma 3D In Vitro Cultures
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benish, B.M. Letter: “The neurocristopathies: A unifying concept of disease arising in neural crest development”. Hum. Pathol. 1975, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef]
- Pudela, C.; Balyasny, S.; Applebaum, M.A. Nervous system: Embryonal tumors: Neuroblastoma. Atlas Genet. Cytogenet Oncol. Haematol. 2020, 24, 284–290. [Google Scholar] [CrossRef]
- Meitar, D.; Crawford, S.E.; Rademaker, A.W.; Cohn, S.L. Tumor angiogenesis correlates with metastatic disease, N-myc amplification, and poor outcome in human neuroblastoma. J. Clin. Oncol. 1996, 14, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Brisse, H.J.; McCarville, M.B.; Granata, C.; Krug, K.B.; Wootton-Gorges, S.L.; Kanegawa, K.; Giammarile, F.; Schmidt, M.; Shulkin, B.L.; Matthay, K.K.; et al. Guidelines for imaging and staging of neuroblastic tumors: Consensus report from the International Neuroblastoma Risk Group Project. Radiology 2011, 261, 243–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikegaki, N.; Shimada, H.; International Neuroblastoma Pathology, C. Subgrouping of Unfavorable Histology Neuroblastomas With Immunohistochemistry Toward Precision Prognosis and Therapy Stratification. JCO Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Naranjo, A.; Irwin, M.S.; Hogarty, M.D.; Cohn, S.L.; Park, J.R.; London, W.B. Statistical Framework in Support of a Revised Children’s Oncology Group Neuroblastoma Risk Classification System. JCO Clin. Cancer Inform. 2018, 2, 1–15. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Janoueix-Lerosey, I.; Ribeiro, A.; Klijanienko, J.; Couturier, J.; Pierron, G.; Mosseri, V.; Valent, A.; Auger, N.; Plantaz, D.; et al. Accumulation of segmental alterations determines progression in neuroblastoma. J. Clin. Oncol. 2010, 28, 3122–3130. [Google Scholar] [CrossRef]
- Schramm, A.; Koster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleveld, T.F.; Schild, L.; Koster, J.; Zwijnenburg, D.A.; Alles, L.K.; Ebus, M.E.; Volckmann, R.; Tijtgat, G.A.; van Sluis, P.; Versteeg, R.; et al. RAS-MAPK Pathway-Driven Tumor Progression Is Associated with Loss of CIC and Other Genomic Aberrations in Neuroblastoma. Cancer Res. 2018, 78, 6297–6307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regairaz, M.; Munier, F.; Sartelet, H.; Castaing, M.; Marty, V.; Renauleaud, C.; Doux, C.; Delbe, J.; Courty, J.; Fabre, M.; et al. Mutation-Independent Activation of the Anaplastic Lymphoma Kinase in Neuroblastoma. Am. J. Pathol. 2016, 186, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, J.H.; Bachmann, H.S.; Brockmeyer, B.; Depreter, K.; Oberthur, A.; Ackermann, S.; Kahlert, Y.; Pajtler, K.; Theissen, J.; Westermann, F.; et al. High ALK receptor tyrosine kinase expression supersedes ALK mutation as a determining factor of an unfavorable phenotype in primary neuroblastoma. Clin. Cancer Res. 2011, 17, 5082–5092. [Google Scholar] [CrossRef] [Green Version]
- Moreno, L.; Barone, G.; DuBois, S.G.; Molenaar, J.; Fischer, M.; Schulte, J.; Eggert, A.; Schleiermacher, G.; Speleman, F.; Chesler, L.; et al. Accelerating drug development for neuroblastoma: Summary of the Second Neuroblastoma Drug Development Strategy forum from Innovative Therapies for Children with Cancer and International Society of Paediatric Oncology Europe Neuroblastoma. Eur. J. Cancer 2020, 136, 52–68. [Google Scholar] [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Pearson, A.D.; Herold, R.; Rousseau, R.; Copland, C.; Bradley-Garelik, B.; Binner, D.; Capdeville, R.; Caron, H.; Carleer, J.; Chesler, L.; et al. Implementation of mechanism of action biology-driven early drug development for children with cancer. Eur. J. Cancer 2016, 62, 124–131. [Google Scholar] [CrossRef]
- George, S.L.; Izquierdo, E.; Campbell, J.; Koutroumanidou, E.; Proszek, P.; Jamal, S.; Hughes, D.; Yuan, L.; Marshall, L.V.; Carceller, F.; et al. A tailored molecular profiling programme for children with cancer to identify clinically actionable genetic alterations. Eur. J. Cancer 2019, 121, 224–235. [Google Scholar] [CrossRef]
- Harttrampf, A.C.; Lacroix, L.; Deloger, M.; Deschamps, F.; Puget, S.; Auger, N.; Vielh, P.; Varlet, P.; Balogh, Z.; Abbou, S.; et al. Molecular Screening for Cancer Treatment Optimization (MOSCATO-01) in Pediatric Patients: A Single-Institutional Prospective Molecular Stratification Trial. Clin. Cancer Res. 2017, 23, 6101–6112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillet, J.P.; Calcagno, A.M.; Varma, S.; Marino, M.; Green, L.J.; Vora, M.I.; Patel, C.; Orina, J.N.; Eliseeva, T.A.; Singal, V.; et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 18708–18713. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. US cancer institute to overhaul tumour cell lines. Nature 2016, 530, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, J.; Valind, A.; Holmquist Mengelbier, L.; Bredin, S.; Cornmark, L.; Jansson, C.; Wali, A.; Staaf, J.; Viklund, B.; Ora, I.; et al. Four evolutionary trajectories underlie genetic intratumoral variation in childhood cancer. Nat. Genet. 2018, 50, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Mengelbier, L.H.; Karlsson, J.; Lindgren, D.; Valind, A.; Lilljebjorn, H.; Jansson, C.; Bexell, D.; Braekeveldt, N.; Ameur, A.; Jonson, T.; et al. Intratumoral genome diversity parallels progression and predicts outcome in pediatric cancer. Nat. Commun. 2015, 6, 6125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, Y.; Tsuchida, Y.; Maseki, N.; Takasaki, N.; Sakurai, M.; Saito, S. Chromosome findings in human neuroblastomas xenografted in nude mice. Jpn. J. Cancer Res. 1985, 76, 359–364. [Google Scholar] [PubMed]
- Tsuchida, Y.; Kaneko, Y.; Kanda, N.; Makino, S.; Utakoji, T.; Saito, S. Possible relationship of chromosome abnormalities and gene amplification with effects of chemotherapy: A neuroblastoma xenograft study. Prog. Clin. Biol. Res. 1985, 175, 171–180. [Google Scholar]
- Tsuchida, Y.; Yokomori, K.; Iwanaka, T.; Saito, S. Nude mouse xenograft study for treatment of neuroblastoma: Effects of chemotherapeutic agents and surgery on tumor growth and cell kinetics. J. Pediatr. Surg. 1984, 19, 72–76. [Google Scholar] [CrossRef]
- Meehan, T.F.; Conte, N.; Goldstein, T.; Inghirami, G.; Murakami, M.A.; Brabetz, S.; Gu, Z.; Wiser, J.A.; Dunn, P.; Begley, D.A.; et al. PDX-MI: Minimal Information for Patient-Derived Tumor Xenograft Models. Cancer Res. 2017, 77, e62–e66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkman, B.E.; Howard, D.; Wendler, D. Reconsidering the Need for Reconsent at 18. Pediatrics 2018, 142. [Google Scholar] [CrossRef] [Green Version]
- Brothers, K.B.; Wilfond, B.S. Research Consent at the Age of Majority: Preferable but not Obligatory. Pediatrics 2018, 142. [Google Scholar] [CrossRef]
- Gastman, B.; Agarwal, P.K.; Berger, A.; Boland, G.; Broderick, S.; Butterfield, L.H.; Byrd, D.; Fecci, P.E.; Ferris, R.L.; Fong, Y.; et al. Defining best practices for tissue procurement in immuno-oncology clinical trials: Consensus statement from the Society for Immunotherapy of Cancer Surgery Committee. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Conde, E.; Angulo, B.; Izquierdo, E.; Paz-Ares, L.; Belda-Iniesta, C.; Hidalgo, M.; Lopez-Rios, F. Lung adenocarcinoma in the era of targeted therapies: Histological classification, sample prioritization, and predictive biomarkers. Clin. Transl. Oncol. 2013, 15, 503–508. [Google Scholar] [CrossRef] [Green Version]
- McDonald, S.A.; Chernock, R.D.; Leach, T.A.; Kahn, A.A.; Yip, J.H.; Rossi, J.; Pfeifer, J.D. Procurement of Human Tissues for Research Banking in the Surgical Pathology Laboratory: Prioritization Practices at Washington University Medical Center. Biopreserv. Biobank. 2011, 9, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Mohseni, M.J.; Amanpour, S.; Muhammadnejad, S.; Sabetkish, S.; Muhammadnejad, A.; Heidari, R.; Haddadi, M.; Mazaheri, Z.; Vasei, M.; Kajbafzadeh, A.M. Establishment of a patient-derived Wilms’ tumor xenograft model: A promising tool for individualized cancer therapy. J. Pediatr. Urol. 2014, 10, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Monsma, D.J.; Cherba, D.M.; Richardson, P.J.; Vance, S.; Rangarajan, S.; Dylewski, D.; Eugster, E.; Scott, S.B.; Beuschel, N.L.; Davidson, P.J.; et al. Using a rhabdomyosarcoma patient-derived xenograft to examine precision medicine approaches and model acquired resistance. Pediatr. Blood Cancer 2014, 61, 1570–1577. [Google Scholar] [CrossRef]
- Wakefield, C.E.; Doolan, E.L.; Fardell, J.E.; Signorelli, C.; Quinn, V.F.; Tucker, K.M.; Patenaude, A.F.; Marshall, G.M.; Lock, R.B.; Georgiou, G.; et al. The Avatar Acceptability Study: Survivor, Parent and Community Willingness to Use Patient-Derived Xenografts to Personalize Cancer Care. EBioMedicine 2018, 37, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Application of Highly Immunocompromised Mice for the Establishment of Patient-Derived Xenograft (PDX) Models. Cells 2019, 8, 889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondarenko, G.; Ugolkov, A.; Rohan, S.; Kulesza, P.; Dubrovskyi, O.; Gursel, D.; Mathews, J.; O’Halloran, T.V.; Wei, J.J.; Mazar, A.P. Patient-Derived Tumor Xenografts Are Susceptible to Formation of Human Lymphocytic Tumors. Neoplasia 2015, 17, 735–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diebel, K.W.; Oko, L.M.; Medina, E.M.; Niemeyer, B.F.; Warren, C.J.; Claypool, D.J.; Tibbetts, S.A.; Cool, C.D.; Clambey, E.T.; van Dyk, L.F. Gammaherpesvirus small noncoding RNAs are bifunctional elements that regulate infection and contribute to virulence in vivo. mBio 2015, 6, e01670-14. [Google Scholar] [CrossRef] [Green Version]
- Hillen, K.M.; Gather, R.; Enders, A.; Pircher, H.; Aichele, P.; Fisch, P.; Blumenthal, B.; Schamel, W.W.; Straub, T.; Goodnow, C.C.; et al. T cell expansion is the limiting factor of virus control in mice with attenuated TCR signaling: Implications for human immunodeficiency. J. Immunol. 2015, 194, 2725–2734. [Google Scholar] [CrossRef] [PubMed]
- Stockmann, C.; Schadendorf, D.; Klose, R.; Helfrich, I. The impact of the immune system on tumor: Angiogenesis and vascular remodeling. Front. Oncol. 2014, 4, 69. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Kamili, A.; Gifford, A.J.; Li, N.; Mayoh, C.; Chow, S.O.; Failes, T.W.; Eden, G.L.; Cadiz, R.; Xie, J.; Lukeis, R.E.; et al. Accelerating development of high-risk neuroblastoma patient-derived xenograft models for preclinical testing and personalised therapy. Br J. Cancer 2020, 122, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Ha, G.; Tseng, Y.Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [Green Version]
- Woo, X.Y.; Giordano, J.; Srivastava, A.; Zhao, Z.M.; Lloyd, M.W.; de Bruijn, R.; Suh, Y.S.; Patidar, R.; Chen, L.; Scherer, S.; et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat. Genet. 2021, 53, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef]
- Roth, A.; Khattra, J.; Yap, D.; Wan, A.; Laks, E.; Biele, J.; Ha, G.; Aparicio, S.; Bouchard-Cote, A.; Shah, S.P. PyClone: Statistical inference of clonal population structure in cancer. Nat. Methods 2014, 11, 396–398. [Google Scholar] [CrossRef]
- Bruna, A.; Rueda, O.M.; Greenwood, W.; Batra, A.S.; Callari, M.; Batra, R.N.; Pogrebniak, K.; Sandoval, J.; Cassidy, J.W.; Tufegdzic-Vidakovic, A.; et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell 2016, 167, 260–274.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callari, M.; Batra, A.S.; Batra, R.N.; Sammut, S.J.; Greenwood, W.; Clifford, H.; Hercus, C.; Chin, S.F.; Bruna, A.; Rueda, O.M.; et al. Computational approach to discriminate human and mouse sequences in patient-derived tumour xenografts. BMC Genom. 2018, 19, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahdesmaki, M.J.; Gray, S.R.; Johnson, J.H.; Lai, Z. Disambiguate: An open-source application for disambiguating two species in next generation sequencing data from grafted samples. F1000Research 2016, 5, 2741. [Google Scholar] [CrossRef] [PubMed]
- Conway, T.; Wazny, J.; Bromage, A.; Tymms, M.; Sooraj, D.; Williams, E.D.; Beresford-Smith, B. Xenome—A tool for classifying reads from xenograft samples. Bioinformatics 2012, 28, i172–i178. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. Patient-derived orthotopic xenografts: Better mimic of metastasis than subcutaneous xenografts. Nat. Rev. Cancer 2015, 15, 451–452. [Google Scholar] [CrossRef]
- Khanna, C.; Jaboin, J.J.; Drakos, E.; Tsokos, M.; Thiele, C.J. Biologically relevant orthotopic neuroblastoma xenograft models: Primary adrenal tumor growth and spontaneous distant metastasis. In Vivo 2002, 16, 77–85. [Google Scholar]
- Braekeveldt, N.; Wigerup, C.; Gisselsson, D.; Mohlin, S.; Merselius, M.; Beckman, S.; Jonson, T.; Borjesson, A.; Backman, T.; Tadeo, I.; et al. Neuroblastoma patient-derived orthotopic xenografts retain metastatic patterns and geno- and phenotypes of patient tumours. Int. J. Cancer 2015, 136, E252–E261. [Google Scholar] [CrossRef] [Green Version]
- Braekeveldt, N.; Wigerup, C.; Tadeo, I.; Beckman, S.; Sanden, C.; Jonsson, J.; Erjefalt, J.S.; Berbegall, A.P.; Borjesson, A.; Backman, T.; et al. Neuroblastoma patient-derived orthotopic xenografts reflect the microenvironmental hallmarks of aggressive patient tumours. Cancer Lett. 2016, 375, 384–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, E.; Federico, S.M.; Chen, X.; Shelat, A.A.; Bradley, C.; Gordon, B.; Karlstrom, A.; Twarog, N.R.; Clay, M.R.; Bahrami, A.; et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 2017, 549, 96–100. [Google Scholar] [CrossRef]
- Stewart, E.; Shelat, A.; Bradley, C.; Chen, X.; Federico, S.; Thiagarajan, S.; Shirinifard, A.; Bahrami, A.; Pappo, A.; Qu, C.; et al. Development and characterization of a human orthotopic neuroblastoma xenograft. Dev. Biol. 2015, 407, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Kroesen, M.; Brok, I.C.; Reijnen, D.; van Hout-Kuijer, M.A.; Zeelenberg, I.S.; Den Brok, M.H.; Hoogerbrugge, P.M.; Adema, G.J. Intra-adrenal murine TH-MYCN neuroblastoma tumors grow more aggressive and exhibit a distinct tumor microenvironment relative to their subcutaneous equivalents. Cancer Immunol. Immunother. 2015, 64, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Teitz, T.; Stanke, J.J.; Federico, S.; Bradley, C.L.; Brennan, R.; Zhang, J.; Johnson, M.D.; Sedlacik, J.; Inoue, M.; Zhang, Z.M.; et al. Preclinical models for neuroblastoma: Establishing a baseline for treatment. PLoS ONE 2011, 6, e19133. [Google Scholar] [CrossRef]
- Maes, W.; Deroose, C.; Reumers, V.; Krylyshkina, O.; Gijsbers, R.; Baekelandt, V.; Ceuppens, J.; Debyser, Z.; Van Gool, S.W. In vivo bioluminescence imaging in an experimental mouse model for dendritic cell based immunotherapy against malignant glioma. J. Neurooncol. 2009, 91, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Hansson, K.; Radke, K.; Aaltonen, K.; Saarela, J.; Manas, A.; Sjolund, J.; Smith, E.M.; Pietras, K.; Pahlman, S.; Wennerberg, K.; et al. Therapeutic targeting of KSP in preclinical models of high-risk neuroblastoma. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Bate-Eya, L.T.; Ebus, M.E.; Koster, J.; den Hartog, I.J.; Zwijnenburg, D.A.; Schild, L.; van der Ploeg, I.; Dolman, M.E.; Caron, H.N.; Versteeg, R.; et al. Newly-derived neuroblastoma cell lines propagated in serum-free media recapitulate the genotype and phenotype of primary neuroblastoma tumours. Eur. J. Cancer 2014, 50, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Barton, J.; Pacey, K.; Jain, N.; Kasia, T.; Edwards, D.; Thevanesan, C.; Straathof, K.; Barone, G.; Anderson, J. Establishment and phenotyping of neurosphere cultures from primary neuroblastoma samples. F1000Research 2019, 8, 823. [Google Scholar] [CrossRef]
- Coulon, A.; Flahaut, M.; Muhlethaler-Mottet, A.; Meier, R.; Liberman, J.; Balmas-Bourloud, K.; Nardou, K.; Yan, P.; Tercier, S.; Joseph, J.M.; et al. Functional sphere profiling reveals the complexity of neuroblastoma tumor-initiating cell model. Neoplasia 2011, 13, 991–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef] [Green Version]
- Fusco, P.; Parisatto, B.; Rampazzo, E.; Persano, L.; Frasson, C.; Di Meglio, A.; Leslz, A.; Santoro, L.; Cafferata, B.; Zin, A.; et al. Patient-derived organoids (PDOs) as a novel in vitro model for neuroblastoma tumours. BMC Cancer 2019, 19, 970. [Google Scholar] [CrossRef]
- Decaesteker, B.; Denecker, G.; Van Neste, C.; Dolman, E.M.; Van Loocke, W.; Gartlgruber, M.; Nunes, C.; De Vloed, F.; Depuydt, P.; Verboom, K.; et al. TBX2 is a neuroblastoma core regulatory circuitry component enhancing MYCN/FOXM1 reactivation of DREAM targets. Nat. Commun. 2018, 9, 4866. [Google Scholar] [CrossRef]
- Braekeveldt, N.; von Stedingk, K.; Fransson, S.; Martinez-Monleon, A.; Lindgren, D.; Axelson, H.; Levander, F.; Willforss, J.; Hansson, K.; Ora, I.; et al. Patient-Derived Xenograft Models Reveal Intratumor Heterogeneity and Temporal Stability in Neuroblastoma. Cancer Res. 2018, 78, 5958–5969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clohessy, J.G.; Pandolfi, P.P. Mouse hospital and co-clinical trial project--from bench to bedside. Nat. Rev. Clin. Oncol. 2015, 12, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Swanton, C. Implications of cancer evolution for drug development. Nat. Rev. Drug Discov. 2017, 16, 441–442. [Google Scholar] [CrossRef]
- Rokita, J.L.; Rathi, K.S.; Cardenas, M.F.; Upton, K.A.; Jayaseelan, J.; Cross, K.L.; Pfeil, J.; Egolf, L.E.; Way, G.P.; Farrel, A.; et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep. 2019, 29, 1675–1689.e9. [Google Scholar] [CrossRef] [PubMed]
- Krytska, K.; Ryles, H.T.; Sano, R.; Raman, P.; Infarinato, N.R.; Hansel, T.D.; Makena, M.R.; Song, M.M.; Reynolds, C.P.; Mosse, Y.P. Crizotinib Synergizes with Chemotherapy in Preclinical Models of Neuroblastoma. Clin. Cancer Res. 2016, 22, 948–960. [Google Scholar] [CrossRef] [Green Version]
- Trigg, R.M.; Lee, L.C.; Prokoph, N.; Jahangiri, L.; Reynolds, C.P.; Amos Burke, G.A.; Probst, N.A.; Han, M.; Matthews, J.D.; Lim, H.K.; et al. The targetable kinase PIM1 drives ALK inhibitor resistance in high-risk neuroblastoma independent of MYCN status. Nat. Commun. 2019, 10, 5428. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Krytska, K.; Larmour, C.E.; Raman, P.; Martinez, D.; Ligon, G.F.; Lillquist, J.S.; Cucchi, U.; Orsini, P.; Rizzi, S.; et al. An antibody-drug conjugate directed to the ALK receptor demonstrates efficacy in preclinical models of neuroblastoma. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Mohlin, S.; Hansson, K.; Radke, K.; Martinez, S.; Blanco-Apiricio, C.; Garcia-Ruiz, C.; Welinder, C.; Esfandyari, J.; O’Neill, M.; Pastor, J.; et al. Anti-tumor effects of PIM/PI3K/mTOR triple kinase inhibitor IBL-302 in neuroblastoma. EMBO Mol. Med. 2019, 11, e10058. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [Green Version]
- Barry, W.E.; Jackson, J.R.; Asuelime, G.E.; Wu, H.W.; Sun, J.; Wan, Z.; Malvar, J.; Sheard, M.A.; Wang, L.; Seeger, R.C.; et al. Activated Natural Killer Cells in Combination with Anti-GD2 Antibody Dinutuximab Improve Survival of Mice after Surgical Resection of Primary Neuroblastoma. Clin. Cancer Res. 2019, 25, 325–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, R.; Moustaki, A.; Norrie, J.L.; Brown, S.; Akers, W.J.; Shirinifard, A.; Dyer, M.A. Interleukin-15 Enhances Anti-GD2 Antibody-Mediated Cytotoxicity in an Orthotopic PDX Model of Neuroblastoma. Clin. Cancer Res. 2019, 25, 7554–7564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Wen, B.; Anton, O.M.; Yao, Z.; Dubois, S.; Ju, W.; Sato, N.; DiLillo, D.J.; Bamford, R.N.; Ravetch, J.V.; et al. IL-15 enhanced antibody-dependent cellular cytotoxicity mediated by NK cells and macrophages. Proc. Natl. Acad. Sci. USA 2018, 115, E10915–E10924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.C.; Aryee, K.E.; Cheng, M.; Kaur, P.; Keck, J.G.; Brehm, M.A. Creation of PDX-Bearing Humanized Mice to Study Immuno-oncology. Methods Mol. Biol. 2019, 1953, 241–252. [Google Scholar] [CrossRef]
- Nguyen, R.; Patel, A.G.; Griffiths, L.M.; Dapper, J.; Stewart, E.A.; Houston, J.; Johnson, M.; Akers, W.J.; Furman, W.L.; Dyer, M.A. Next-generation humanized patient-derived xenograft mouse model for pre-clinical antibody studies in neuroblastoma. Cancer Immunol. Immunother. 2020, 70, 721–732. [Google Scholar] [CrossRef]
- Whiteford, C.C.; Bilke, S.; Greer, B.T.; Chen, Q.; Braunschweig, T.A.; Cenacchi, N.; Wei, J.S.; Smith, M.A.; Houghton, P.; Morton, C.; et al. Credentialing preclinical pediatric xenograft models using gene expression and tissue microarray analysis. Cancer Res. 2007, 67, 32–40. [Google Scholar] [CrossRef] [Green Version]
- El-Hoss, J.; Jing, D.; Evans, K.; Toscan, C.; Xie, J.; Lee, H.; Taylor, R.A.; Lawrence, M.G.; Risbridger, G.P.; MacKenzie, K.L.; et al. A single nucleotide polymorphism genotyping platform for the authentication of patient derived xenografts. Oncotarget 2016, 7, 60475–60490. [Google Scholar] [CrossRef]
- Townsend, E.C.; Murakami, M.A.; Christodoulou, A.; Christie, A.L.; Koster, J.; DeSouza, T.A.; Morgan, E.A.; Kallgren, S.P.; Liu, H.; Wu, S.C.; et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell 2016, 30, 183. [Google Scholar] [CrossRef]
- Wrobel, J.K.; Najafi, S.; Ayhan, S.; Gatzweiler, C.; Krunic, D.; Ridinger, J.; Milde, T.; Westermann, F.; Peterziel, H.; Meder, B.; et al. Rapid In Vivo Validation of HDAC Inhibitor-Based Treatments in Neuroblastoma Zebrafish Xenografts. Pharmaceuticals 2020, 13, 345. [Google Scholar] [CrossRef]
- Yan, C.; Brunson, D.C.; Tang, Q.; Do, D.; Iftimia, N.A.; Moore, J.C.; Hayes, M.N.; Welker, A.M.; Garcia, E.G.; Dubash, T.D.; et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell 2019, 177, 1903–1914.e14. [Google Scholar] [CrossRef]
- Ikonomopoulou, M.P.; Fernandez-Rojo, M.A.; Pineda, S.S.; Cabezas-Sainz, P.; Winnen, B.; Morales, R.A.V.; Brust, A.; Sanchez, L.; Alewood, P.F.; Ramm, G.A.; et al. Gomesin inhibits melanoma growth by manipulating key signaling cascades that control cell death and proliferation. Sci. Rep. 2018, 8, 11519. [Google Scholar] [CrossRef] [Green Version]
- Eden, C.J.; Ju, B.; Murugesan, M.; Phoenix, T.N.; Nimmervoll, B.; Tong, Y.; Ellison, D.W.; Finkelstein, D.; Wright, K.; Boulos, N.; et al. Orthotopic models of pediatric brain tumors in zebrafish. Oncogene 2015, 34, 1736–1742. [Google Scholar] [CrossRef] [Green Version]
- Kirchberger, S.; Sturtzel, C.; Pascoal, S.; Distel, M. Quo natas, Danio?-Recent Progress in Modeling Cancer in Zebrafish. Front. Oncol. 2017, 7, 186. [Google Scholar] [CrossRef]
- Graham, M.L.; Prescott, M.J. The multifactorial role of the 3Rs in shifting the harm-benefit analysis in animal models of disease. Eur. J. Pharmacol. 2015, 759, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frith, T.J.R.; Tsakiridis, A. Efficient Generation of Trunk Neural Crest and Sympathetic Neurons from Human Pluripotent Stem Cells Via a Neuromesodermal Axial Progenitor Intermediate. Curr. Protoc. Stem Cell Biol. 2019, 49, e81. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Miller, M.L.; McHenry, L.K.; Zheng, T.; Zhen, Q.; Ilkhanizadeh, S.; Conklin, B.R.; Bronner, M.E.; Weiss, W.A. Generating trunk neural crest from human pluripotent stem cells. Sci. Rep. 2016, 6, 19727. [Google Scholar] [CrossRef] [Green Version]
- Menendez, L.; Kulik, M.J.; Page, A.T.; Park, S.S.; Lauderdale, J.D.; Cunningham, M.L.; Dalton, S. Directed differentiation of human pluripotent cells to neural crest stem cells. Nat. Protoc. 2013, 8, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.R.; Otero, J.H.; Garcia-Lopez, J.; Wallace, K.; Finkelstein, D.; Rehg, J.E.; Yin, Z.; Wang, Y.D.; Freeman, K.W. MYCN induces neuroblastoma in primary neural crest cells. Oncogene 2017, 36, 5075–5082. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Peng, Y.; Li, H.; Chen, W. Organ-on-a-Chip: A New Paradigm for Drug Development. Trends Pharmacol. Sci. 2021, 42, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Ma, C.; Vasudevaraja, V.; Serrano, J.; Tong, J.; Peng, Y.; Delorenzo, M.; Shen, G.; Frenster, J.; Morales, R.T.; et al. Dissecting the immunosuppressive tumor microenvironments in Glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tucker, E.R.; George, S.; Angelini, P.; Bruna, A.; Chesler, L. The Promise of Patient-Derived Preclinical Models to Accelerate the Implementation of Personalised Medicine for Children with Neuroblastoma. J. Pers. Med. 2021, 11, 248. https://doi.org/10.3390/jpm11040248
Tucker ER, George S, Angelini P, Bruna A, Chesler L. The Promise of Patient-Derived Preclinical Models to Accelerate the Implementation of Personalised Medicine for Children with Neuroblastoma. Journal of Personalized Medicine. 2021; 11(4):248. https://doi.org/10.3390/jpm11040248
Chicago/Turabian StyleTucker, Elizabeth R., Sally George, Paola Angelini, Alejandra Bruna, and Louis Chesler. 2021. "The Promise of Patient-Derived Preclinical Models to Accelerate the Implementation of Personalised Medicine for Children with Neuroblastoma" Journal of Personalized Medicine 11, no. 4: 248. https://doi.org/10.3390/jpm11040248