
Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: Selective Targeting Cancer Cell Mitochondria

 ,
,  ,
,  and
and 
Abstract
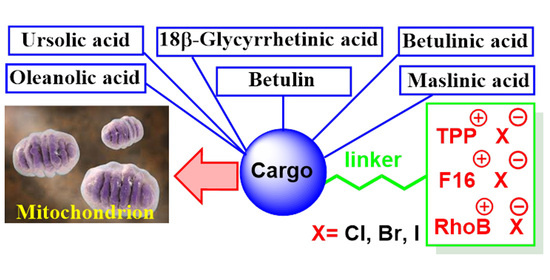
:
1. Introduction
2. Metabolic and Mitochondrial Changes in Cancer Cells
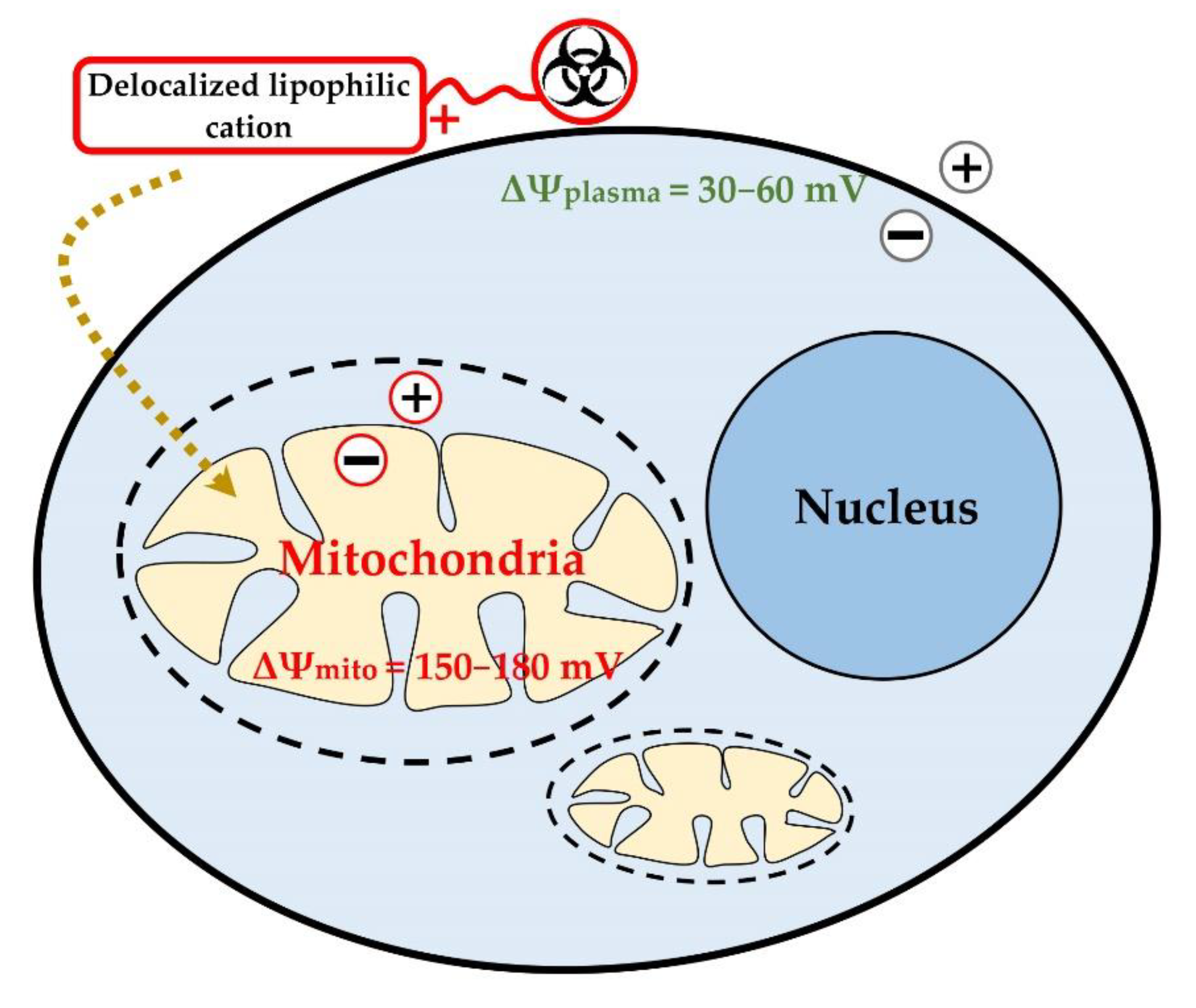
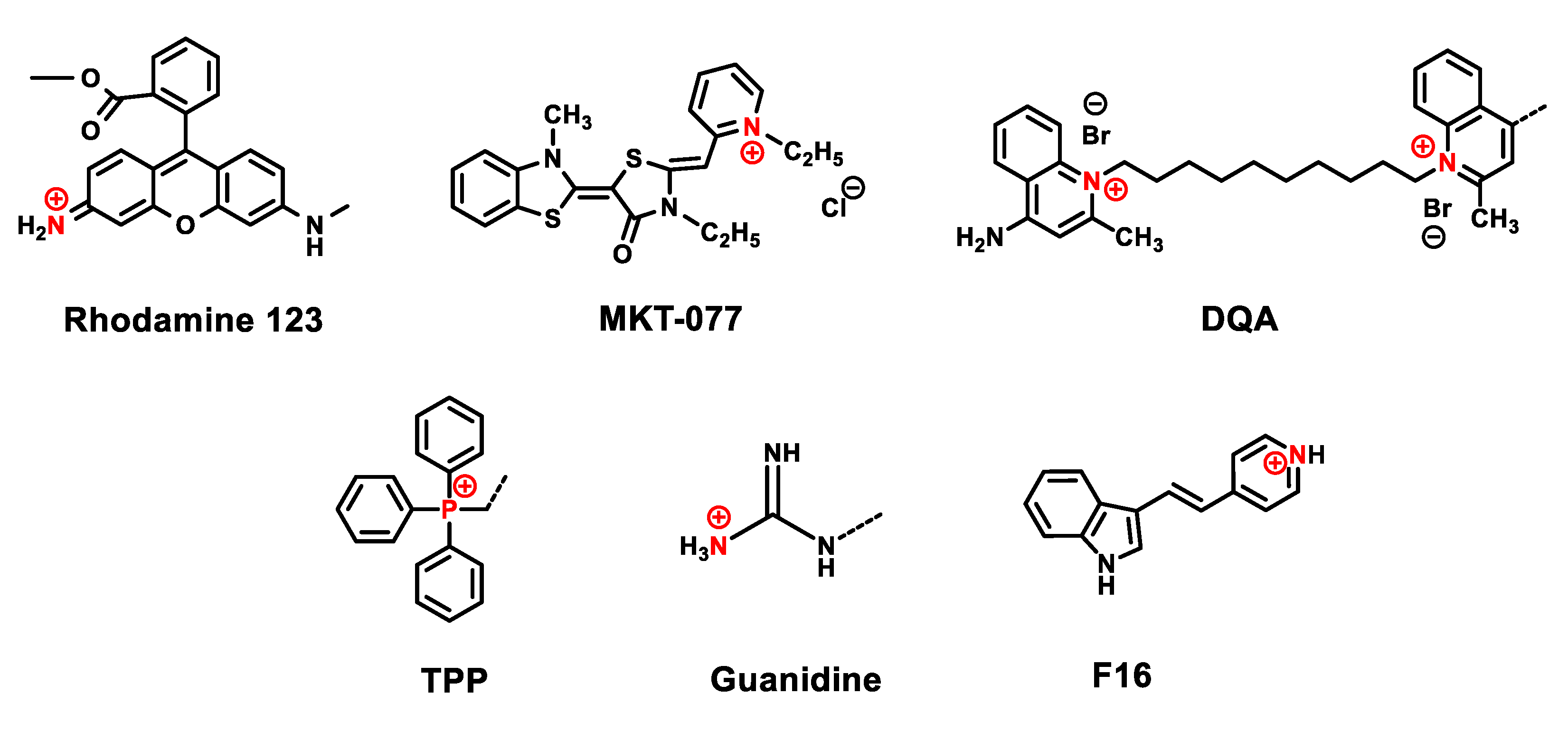
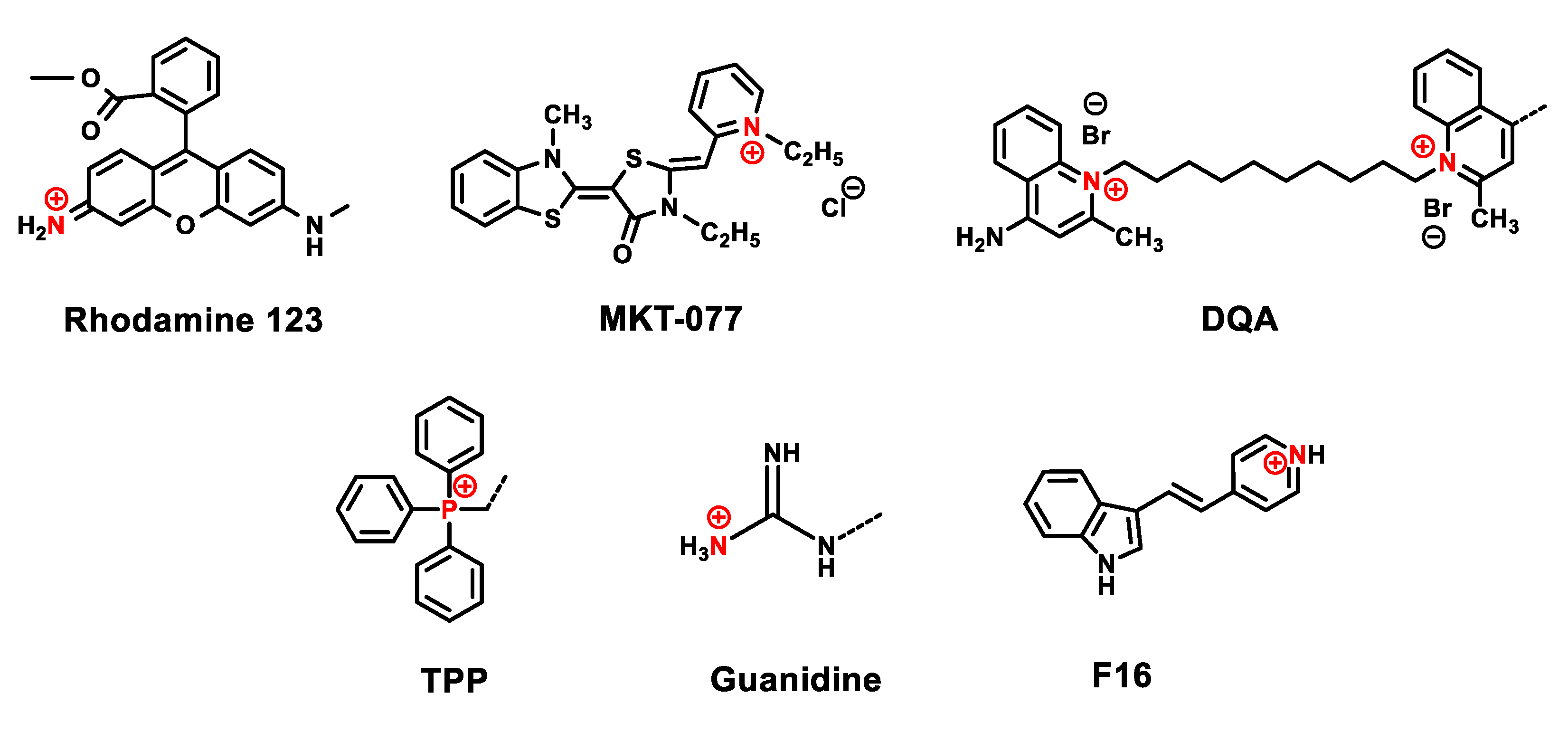
3. Delocalized Lipophilic Cations (DLCs) for Mitochondria-Targeted Drug Delivery
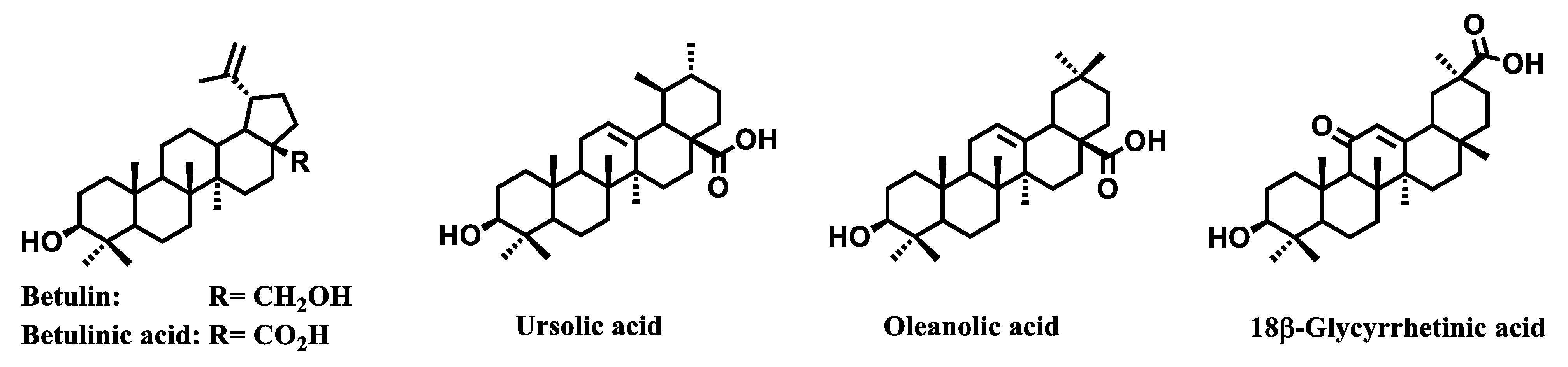
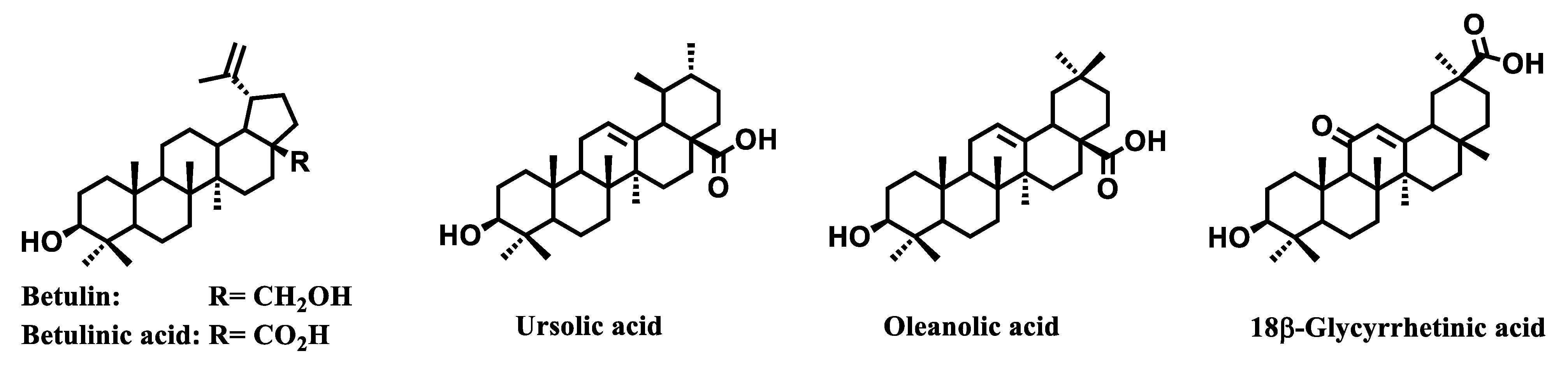
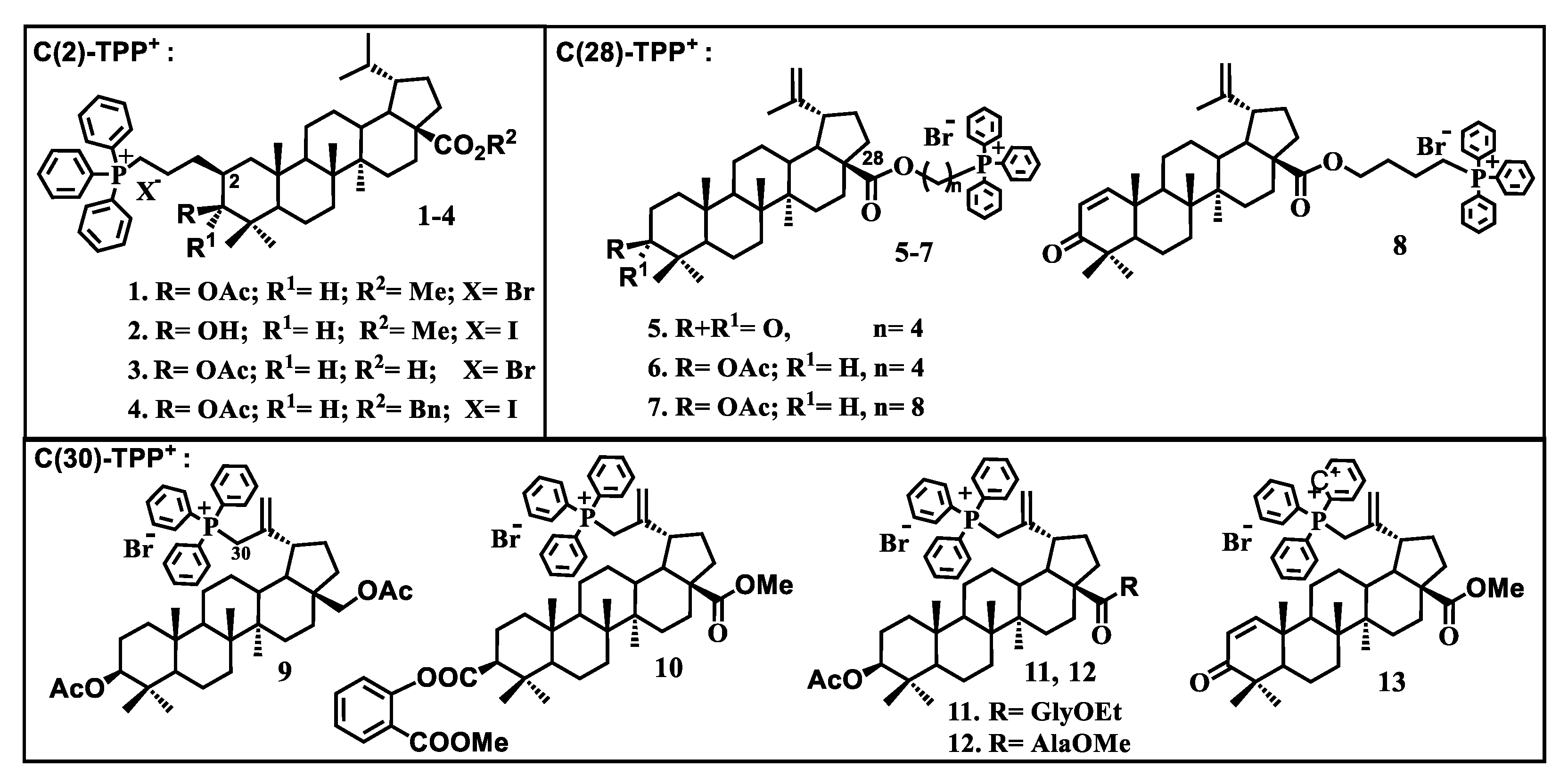
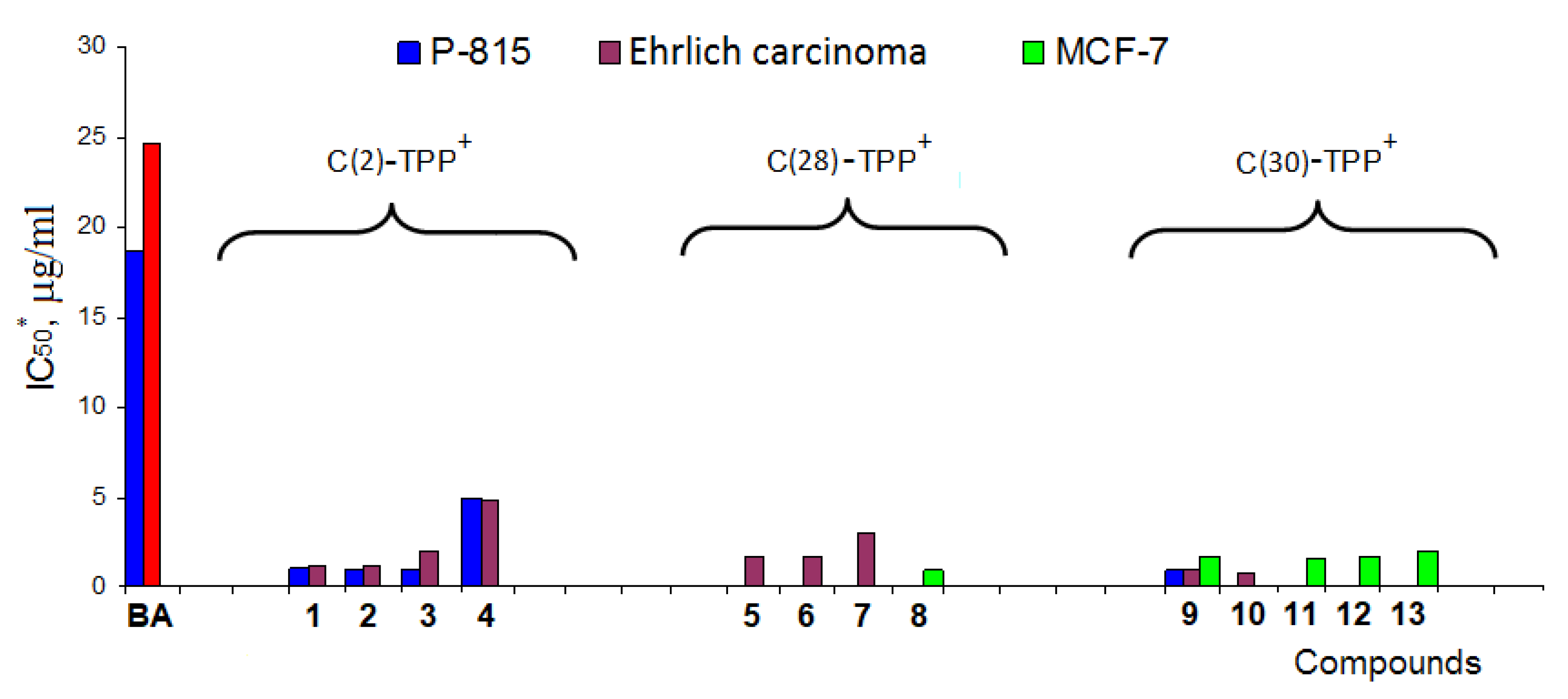
4. Mitochondria-Targeted Conjugates of Triterpenic Acids with DLCs as a Novel Group of Mitocans
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardillos, A.M.; Chiva, A.V.; Vargas, G.B.; Blanco, P.H.; Cid, R.P.; Guijarro, P.J.; Hümmer, S.; Serrano, E.B.; Casanova, A.R.; Lagares, A.D.; et al. The role of clonal communication and heterogeneity in breast cancer. MC Cancer 2019, 19, 666. [Google Scholar]
- Cajal, S.R.Y.; Sese, M.; Capdevila, C.; Aasen, T.; Arruda, L.D.M.; Cano, S.J.D.; Losa, J.H.; Castellvi, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef] [Green Version]
- Alison, M.R.; Lim, S.M.; Nicholson, L.J. Cancer stem cells: Problems for therapy? J. Pathol. 2011, 223, 147–161. [Google Scholar] [CrossRef]
- Neuzil, J.; Tomasetti, M.; Zhao, Y.; Dong, L.-F.; Birringer, M.; Wang, X.-F.; Low, P.; Wu, K.; Salvatore, B.A.; Ralph, S.J. Vitamin E analogs, a novel group of “mitocans,” as anticancer agents: The importance of being redox-silent. Mol. Pharm. 2007, 71, 1185–1199. [Google Scholar] [CrossRef] [Green Version]
- Neuzil, J.; Dong, L.-F.; Ramanathapuram, L.; Hahn, T.; Chladova, M.; Wang, X.-F.; Zobalova, R.; Prochazka, L.; Gold, M.; Freeman, R.; et al. Vitamin E analogues as a novel group of mitocans: Anti-cancer agents that act by targeting mitochondria. Mol. Asp. Med. 2007, 28, 607–645. [Google Scholar] [CrossRef]
- Neuzil, J.; Dong, L.-F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Weissig, V. Treatment strategies that enhance the efficacy and selectivity of mitochondria-targeted anticancer agents. Int. J. Mol. Sci. 2015, 16, 17394–17421. [Google Scholar] [CrossRef] [Green Version]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef] [PubMed]
- Csuk, R. Betulinic acid and its derivatives: A patent review (2008–2013). Expert Opin. Ther. Pat. 2014, 24, 913–923. [Google Scholar] [CrossRef]
- Pathak, A.K.; Bhutani, M.; Nair, A.S.; Ahn, K.S.; Chakraborty, A.; Kadara, H.; Guha, S.; Sethi, G.; Aggarwal, B.B. Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Clin. Cancer Res. 2007, 5, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Wen, X.; Sun, H. Oleanolic acid derivatives for pharmaceutical use: A patent review. Expert Opin. Ther. Pat. 2016, 26, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Green, I.R.; Ali, I.; Khan, I.A.; Ali, Z.; Al-Sadi, A.M.; Ahmed, I. Ursolic acid derivatives for pharmaceutical use: A patent review (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 1061. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, J.; Chen, Y. Betulinic acid and the pharmacological effects of tumor suppression (Review). Mol. Med. Rep. 2016, 14, 4489–4495. [Google Scholar] [CrossRef] [Green Version]
- Damle, A.A.; Pawar, Y.P.; Narkar, A.A. Anticancer activity of betulinic acid on MCF-7 tumors in nude mice. Indian J. Exp. Biol. 2013, 51, 485–491. [Google Scholar]
- Mullauer, F.B.; Bloois, L.; Daalhuisen, J.B.; Brink, M.S.T.; Storm, G.; Medema, J.P.; Schiffelers, R.M.; Kessler, J.H. Betulinic acid delivered in liposomes reduces growth of human lung and colon cancers in mice without causing systemic toxicity. Anticancer Drugs 2011, 22, 223–233. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2020, 38, 107409. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.K.; Dash, S.S.; Chattopadhyay, S.; Ghosh, T.; Tripathy, S.; Kar Mahapatra, S.; Bag, B.G.; Das, D.; Roy, S. Folate decorated delivery of self assembled betulinic acid nanofibers: A biocompatible anti-leukemic therapy. RSC Adv. 2015, 5, 24144–24157. [Google Scholar] [CrossRef]
- Warburg, O. Versuche an überlebendem Carcinomgewebe. Biochem. Zschr. 1923, 142, 317–333. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Asp. Med. 2010, 31, 1–20. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Dubinin, M.V.; Talanov, E.Y.; Starinets, V.S.; Tenkov, K.S.; Zakharova, N.M.; Belosludtseva, N.V. Transport of Ca2+ and Ca2+-Dependent Permeability Transition in the Liver and Heart Mitochondria of Rats with Different Tolerance to Acute Hypoxia. Biomolecules 2020, 10, 114. [Google Scholar] [CrossRef] [Green Version]
- Belosludtsev, K.N.; Talanov, E.Y.; Starinets, V.S.; Agafonov, A.V.; Dubinin, M.V.; Belosludtseva, N.V. Transport of Ca2+ and Ca2+-Dependent Permeability Transition in Rat Liver Mitochondria under the Streptozotocin-Induced Type I Diabetes. Cells 2019, 8, 1014. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Catalán, M.; Olmedo, I.; Faúndez, J.; Jara, J.A. Medicinal chemistry targeting mitochondria: From new vehicles and pharmacophore groups to old drugs with mitochondrial activity. Int. J. Mol. Sci. 2020, 21, 8684. [Google Scholar] [CrossRef]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [Green Version]
- Goto, M.; Miwa, H.; Shikami, M.; Tsunekawa-Imai, N.; Suganuma, K.; Mizuno, S.; Takahashi, M.; Mizutani, M.; Hanamura, I.; Nitta, M. Importance of glutamine metabolism in leukemia cells by energy production through TCA cycle and by redox homeostasis. Cancer Investig. 2014, 32, 241–247. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting aspartate biosynthesis is an essential function of respiration in proliferating xells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Modica-Napolitano, J.S.; Aprille, J.R. Basis for the selective cytotoxicity of rhodamine 123. Cancer Res. 1987, 47, 4361–4365. [Google Scholar]
- Kamo, N.; Muratsugu, M.; Hongoh, R.; Kobatake, Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J. Membr. Biol. 1979, 49, 105–121. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar]
- Baggetto, L.G.; Testa-Parussini, R. Role of acetoin on the regulation of intermediate metabolism of Ehrlich ascites tumor mitochondria: Its contribution to membrane cholesterol enrichment modifying passive proton permeability. Arch. Biochem. Biophys. 1990, 283, 241–248. [Google Scholar] [CrossRef]
- Baggetto, L.G.; Clottes, E.; Vial, C. Low mitochondrial proton leak due to high membrane cholesterol content and cytosolic creatine kinase as two features of the deviant bioenergetics of Ehrlich and AS30-D tumor cells. Cancer Res. 1992, 52, 4935–4941. [Google Scholar] [PubMed]
- Battogtokha, G.; Choia, Y.S.; Kang, D.S.; Park, S.J.; Shim, M.S.; Huh, K.M.; Choa, Y.-Y.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: Current strategies and future perspectives. Acta Pharm. Sin. B 2018, 8, 862–880. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Xiao, Y.; Fu, B.; Qin, Z. TPP-based mitocans: A potent strategy for anticancer drug design. RSC Med. Chem. 2020, 11, 858–875. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Harris, I.; Mak, T. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug. Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sánchez, R. The causes of cancer revisited: “mitochondrial malignancy” and ROS-induced oncogenic transformation-why mitochondria are targets for cancer therapy. Mol. Asp. Med. 2010, 31, 145–170. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Bajzikova, M.; Kovarova, J.; Coelho, A.R.; Boukalova, S.; Oh, S.; Rohlenova, K.; Svec, D.; Hubackova, S.; Endaya, B.; Judasova, K.; et al. Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab. 2019, 29, 399–416.e10. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.Q.; Zhang, X.; Zhang, S.; Zhu, T.; Garg, M.; Lobie, P.E.; Pandey, V. Mitochondria: The metabolic switch of cellular oncogenic transformation. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188534. [Google Scholar] [CrossRef]
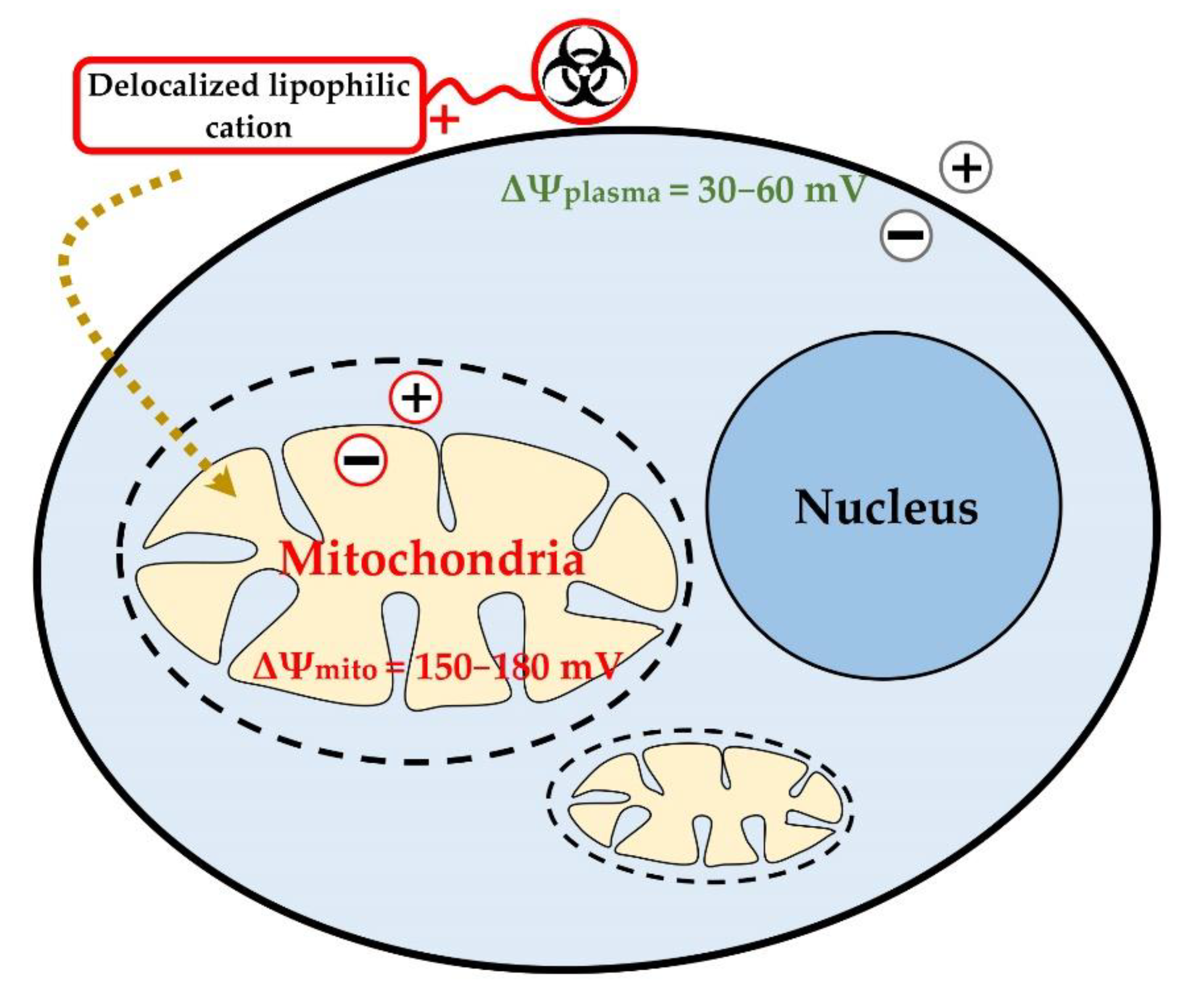
- Modica-Napolitano, J.S.; Aprille, J.R. Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv. Drug Deliv. Rev. 2001, 49, 63–70. [Google Scholar] [CrossRef]
- Wang, F.; Ogasawara, M.A.; Huang, P. Small mitochondria-targeting molecules as anti-cancer agents. Mol. Asp. Med. 2010, 31, 75–92. [Google Scholar] [CrossRef] [Green Version]
- Fantin, V.R.; Berardi, M.J.; Scorrano, L.; Korsmeyer, S.J.; Leder, P. A novel mitochondriotoxic small molecule that selectively inhibits tumor cell growth. Cancer Cell 2002, 2, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Ross, M.F.; Kelso, G.F.; Blaikie, F.H.; James, A.M.; Cocheme, H.M.; Filipovska, A.; Ros, T.; Hurd, T.R.; Smith, R.A.J.; Murphy, M.P. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry 2005, 70, 222–230. [Google Scholar] [CrossRef]
- Weiss, M.J.; Wong, J.R.; Ha, C.S.; Bleday, R.; Salem, R.R.; Steele, G.D., Jr.; Chen, L.B. Dequalinium, a topical antimicrobial agent, displays anticarcinoma activity based on selective mitochondrial accumulation. Proc. Natl. Acad. Sci. USA 1987, 84, 5444–5448. [Google Scholar] [CrossRef] [Green Version]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Tenkov, K.S.; Sharapov, V.A.; Kosareva, E.A.; Dubinin, M.V. Effect of Dequalinium on Respiration and the Inner Membrane Permeability of Rat Liver Mitochondria. Biochem. Suppl. Ser. A Membr. Cell Biol. 2018, 12, 121–127. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Weiss, M.J.; Chen, L.B.; Aprille, J.R. Rhodamine 123 inhibits bioenergetic function in isolated rat liver mitochondria. Biochem. Biophys. Res. Commun. 1984, 118, 717–723. [Google Scholar] [CrossRef]
- Fantin, V.R.; Leder, P. F16, a mitochondriotoxic compound, triggers apoptosis or necrosis depending on the genetic background of the target carcinoma cell. Cancer Res. 2004, 64, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Koya, K.; Li, Y.; Wang, H.; Ukai, T.; Tatsuta, N.; Kawakami, M.; Shishido, T.; Chen, L.B. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996, 56, 538–543. [Google Scholar]
- Modica-Napolitano, J.S.; Koya, K.; Weisberg, E.; Brunelli, B.T.; Li, Y.; Chen, L.B. Selective damage to carcinoma mitochondria by the rhodacyanine MKT-077. Cancer Res. 1996, 56, 544–550. [Google Scholar] [PubMed]
- Britten, C.D.; Rowinsky, E.K.; Baker, S.D.; Weiss, G.R.; Smith, L.; Stephenson, J.; Rothenberg, M.; Smetzer, L.; Cramer, J.; Collins, W.; et al. Eckhardt, S.G. A phase I and pharmacokinetic study of the mitochondrial-specific rhodacyanine dye analog MKT 077. Clin. Cancer Res. 2000, 6, 42–49. [Google Scholar]
- Propper, D.J.; Braybrooke, J.P.; Taylor, D.J.; Lodi, R.; Styles, P.; Cramer, J.A.; Collins, W.C.J.; Levitt, N.C.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I trial of the selective mitochondrial toxin MKT 077 in chemo-resistant solid tumours. Ann. Oncol. 1999, 10, 923–927. [Google Scholar] [CrossRef]
- Skulachev, V.P. A biochemical approach to the problem of aging: “Megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry 2007, 72, 1385–1396. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P.; Anisimov, V.N.; Antonenko, Y.N.; Bakeeva, L.E.; Chernyak, B.V.; Erichev, V.P.; Filenko, O.F.; Kalinina, N.I.; Kapelko, V.I.; Kolosova, N.G.; et al. An attempt to prevent senescence: A mitochondrial approach. Biochim. Biophys. Acta 2009, 1787, 437–461. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. Cationic antioxidants as a powerful tool against mitochondrial oxidative stress. Biochem. Biophys. Res. Commun. 2013, 441, 275–279. [Google Scholar] [CrossRef]
- Jauslin, M.L.; Meier, T.; Smith, R.A.J.; Murphy, M.P. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003, 17, 1972–1974. [Google Scholar] [CrossRef]
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; Arroyo-Jimenez, M.D.M.; Jordán, J.; Galindo, M.F. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim. Biophys. Acta 2013, 1832, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Stantic, M.; Zobalova, R.; Bezawork-Geleta, A.; Stapelberg, M.; Stursa, J.; Prokopova, K.; Dong, L.; Neuzil, J. Mitochondrially targeted vitamin E succinate efficiently kills breast tumour-initiating cells in a complex II-dependent manner. BMC Cancer 2015, 15, 401. [Google Scholar] [CrossRef] [Green Version]
- Millard, M.; Gallagher, J.D.; Olenyuk, B.Z.; Neamati, N. A Selective Mitochondrial-Targeted Chlorambucil with Remarkable Cytotoxicity in Breast and Pancreatic Cancers. J. Med. Chem. 2013, 22, 9170–9179. [Google Scholar] [CrossRef]
- Han, M.; Vakili, M.R.; Abyaneh, H.S.; Molavi, O.; Lai, R.; Lavasanifar, A. Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX cells. Mol. Pharm. 2014, 11, 2640–2649. [Google Scholar] [CrossRef]
- Boukalova, S.; Stursa, J.; Werner, L.; Ezrova, Z.; Cerny, J.; Bezawork-Geleta, A.; Pecinova, A.; Dong, L.; Drahota, Z.; Neuzil, J. Mitochondrial Targeting of Metformin Enhances Its Activity against Pancreatic Cancer. Mol. Cancer 2016, 15, 2875–2886. [Google Scholar] [CrossRef] [Green Version]
- Rohlenova, K.; Sachaphibulkij, K.; Stursa, J.; Bezawork-Geleta, A.; Blecha, J.; Endaya, B.; Werner, L.; Cerny, J.; Zobalova, R.; Goodwin, J.; et al. Selective Disruption of Respiratory Supercomplexes as a New Strategy to Suppress Her2high Breast Cancer. Antioxid. Redox Signal. 2017, 26, 84–103. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.; Swargiary, G.; Singh, K.K. Natural Agents Targeting Mitochondria in Cancer. Int. J. Mol. Sci. 2020, 21, 6992. [Google Scholar] [CrossRef]
- Dong, L.-F.; Jameson, V.J.A.; Tilly, D.; Cerny, J.; Mahdavian, E.; Marín-Herna´ndez, A.; Herna´ndez-Esquivel, L.; Rodríguez-Enríquez, S.; Stursa, J.; Witting, P.K.; et al. Mitochondrial Targeting of Vitamin E Succinate Enhances Its Pro-apoptotic and Anti-cancer Activity via Mitochondrial Complex II. J. Biol. Chem. 2011, 5, 3717–3728. [Google Scholar] [CrossRef] [Green Version]
- Rohlena, J.; Dong, L.-F.; Kluckova, K.; Zobalova, R.; Goodwin, J.; Tilly, D.; Stursa, J.; Pecinova, A.; Philimonenko, A.; Hozak, P.; et al. Mitochondrially Targeted a-Tocopheryl Succinate Is Antiangiogenic: Potential Benefit Against Tumor Angiogenesis but Caution Against Wound Healing. Antioxid. Redox Signal. 2011, 15, 2923–2935. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.-F.; Low, P.; Dyason, J.C.; Wang, X.-F.; Prochazka, L.; Witting, P.K.; Freeman, R.; Swettenham, E.; Valis, K.; Liu, J.; et al. α-Tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 2008, 27, 4324–4335. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Mu, X.; You, Q. Succinate: An initiator in tumorigenesis and progression. Oncotarget 2017, 8, 53819–53828. [Google Scholar] [CrossRef] [Green Version]
- Wilken, R.; Veena, M.S.; Wang, M.B.; Srivatsan, E.S. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol. Cancer 2011, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Reddy, C.A.; Somepalli, V.; Golakoti, T.; Kanugula, A.K.; Karnewar, S.; Rajendiran, K.; Vasagiri, N.; Prabhakar, S.; Kuppusamy, P.; Kotamraju, S.; et al. Mitochondrial-targeted curcuminoids: A strategy to enhance bioavailability and anticancer efficacy of curcumin. PLoS ONE 2014, 9, e89351. [Google Scholar] [CrossRef] [Green Version]
- Cichewicz, R.H.; Kouzi, S.A. Chemistry, biological activity, and chemotherapeutic potential of betulinic acid for the prevention and treatment of cancer and HIV infection. Med. Res. Rev. 2004, 24, 90–114. [Google Scholar] [CrossRef]
- Sarek, J.; Kvasnica, M.; Vlk, M.; Urban, M.; Dzubak, P.; Hajduch, M. Research on Melanoma: A Glimpse into Current Directions and Future Trends; Murph, M., Ed.; In Tech: Rijeka, Croatia, 2011; Volume 7, p. 125. [Google Scholar]
- Mukherjee, R.; Kumar, V.; Srivastava, S.K.; Agarwal, S.K.; Burman, A.C. Betulinic acid derivatives as anticancer agents: Structure activity relationship. Anti Cancer Agents Med. Chem. 2006, 6, 271–279. [Google Scholar] [CrossRef]
- Ali-Seyed, M.; Jantan, I.; Vijayaraghavan, K.; Bukhari, S.N.A. Betulinic Acid: Recent Advances in Chemical Modifications, Effective Delivery, and Molecular Mechanisms of a Promising Anticancer Therapy. Chem. Biol. Drug Des. 2016, 87, 517–536. [Google Scholar] [CrossRef]
- Zhang, D.-M.; Xu, H.-G.; Wang, L.; Li, Y.-J.; Sun, P.-H.; Wu, X.-M.; Wang, G.-J.; Chen, W.-M.; Ye, W.-C. Betulinic Acid and its Derivatives as Potential Antitumor Agents. Med. Res. Rev. 2015, 35, 1127–1155. [Google Scholar] [CrossRef]
- Wang, X.; Lu, X.; Zhu, R.; Zhang, K.; Li, S.; Chen, Z.; Li, L. Betulinic acid induces apoptosis in differentiated PC12 cells Via ROS-mediated mitochondrial pathway. Neurochem. Res. 2017, 42, 1130–1140. [Google Scholar] [CrossRef]
- Tan, Y.M.; Yu, R.; Pezzuto, J.M. Betulinic acid-induced programmed cell death in human melanoma cells involves mitogen-activated protein kinase activation. Clin. Cancer Res. 2003, 9, 2866–2875. [Google Scholar]
- Fulda, S.; Kroemer, G. Targeting mitochondrial apoptosis by betulinic acid in human cancers. Drug Discov. Today 2009, 14, 885–890. [Google Scholar] [CrossRef]
- Fulda, S.; Kroemer, G. Mitochondria as therapeutic targets for the treatment of malignant disease. Antioxid. Redox Signal. 2011, 15, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Shishodia, S.; Majumdar, S.; Banerjee, S.; Aggarwal, B.B. Ursolic acid inhibits nuclear factor-κB activation induced by carcinogenic agents through suppression of IκBα kinase and p65 phosphorylation: Correlation with down-regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1. Cancer Res. 2003, 63, 4375–4383. [Google Scholar] [PubMed]
- Chen, H.; Gao, Y.; Wang, A.; Zhou, X.; Zheng, Y.; Zhou, J. Evolution in medicinal chemistry of ursolic acid derivatives as anticancer agents. Eur. J. Med. Chem. 2015, 92, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Villar, V.H.; Vögler, O.; Barceló, F.; Broto, J.M.; Serra, J.M.; Gutiérrez, V.R.; Alemany, R. Down-regulation of AKT signalling by ursolic acid induces intrinsic apoptosis and sensitization to doxorubicin in soft tissue sarcoma. PLoS ONE 2016, 11, e0155946. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Dai, X.; Kumar, A.P.; Tan, B.K.H.; Sethi, G.; Bishayee, A. Ursolic acid in cancer prevention and treatment: Molecular targets, pharmacokinetics and clinical studies. Biochem. Pharm. 2013, 85, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Semenova, A.A.; Ilzorkina, A.I.; Mikheeva, I.B.; Yashin, V.A.; Penkov, N.V.; Vydrina, V.A.; Ishmuratov, G.Y.; Sharapov, V.A.; Khoroshavina, E.I.; et al. Effect of betulin and betulonic acid on isolated rat liver mitochondria and liposomes. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183383. [Google Scholar] [CrossRef]
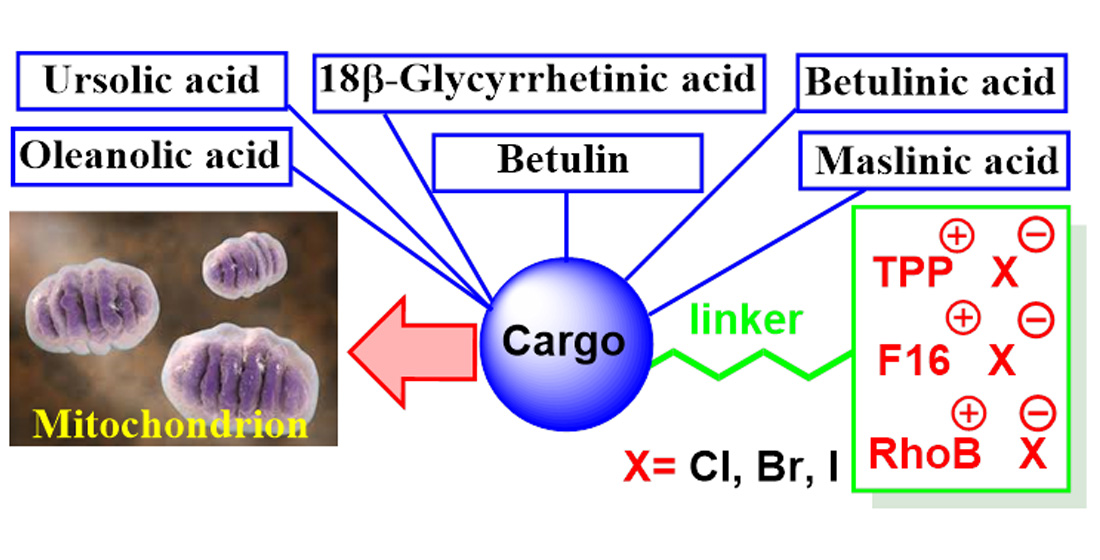
- Spivak, A.Y.; Nedopekina, D.A.; Shakurova, E.R.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Dzhemilev, U.M.; Bel’skii, Y.P.; Bel’skaya, N.V.; Stankevich, S.A.; et al. Synthesis of lupane triterpenoids with triphenylphosphonium substituents and studies of their antitumor activity. Russ. Chem. Bull. 2013, 62, 188–198. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Bel’skii, Y.P.; Bel’skaya, N.V.; Khazanov, V.A. Triphenylphosphonium cations of betulinic acid derivatives: Synthesis and antitumor activity. Med. Chem. Res. 2017, 26, 518–531. [Google Scholar] [CrossRef]
- Tsepaeva, O.V.; Nemtarev, A.V.; Abdullin, T.I.; Grigor’eva, L.R.; Kuznetsova, E.V.; Akhmadishina, R.A.; Ziganshina, L.E.; Cong, H.H.; Mironov, V.F. Design, Synthesis, and Cancer Cell Growth Inhibitory Activity of Triphenylphosphonium Derivatives of the Triterpenoid Betulin. J. Nat. Prod. 2017, 80, 2232–2239. [Google Scholar] [CrossRef]
- Nedopekina, D.A.; Gubaidullin, R.R.; Odinokov, V.N.; Maximchik, P.V.; Zhivotovsky, B.; Bel’skii, Y.P.; Khazanov, V.A.; Manuylova, A.V.; Gogvadze, V.; Spivak, A.Y. Mitohondria-targeted betulinic and ursolic acid derivatives: Synthesis and anticancer activity. Med. Chem. Commun. 2017, 8, 1934–1945. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Dai, L.; Ji, M.; Wang, H. Mitochondria-targeted triphenylphosphonium conjugated glycyrrhetinic acid derivatives as potent anticancer drugs. Bioorg. Chem. 2019, 85, 179–190. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, T.; Yuan, H.; Li, D.; Lou, H.; Fan, P. Mitochondria-Targeted Lupane Triterpenoid Derivatives and Their Selective Apoptosis-Inducing Anticancer Mechanisms. J. Med. Chem. 2017, 60, 6353–6363. [Google Scholar] [CrossRef]
- Wang, J.; Fan, X.Y.; Yang, L.Y.; He, H.; Huang, R.; Jiang, F.L.; Liu, Y. Conjugated 5-fluorouracil with mitochondria-targeting lipophilic cation: Design, synthesis and biological evaluation. Med. Chem. Commun. 2016, 7, 2016–2019. [Google Scholar] [CrossRef]
- Peng, Y.B.; Zhao, Z.L.; Liu, T.; Xie, G.J.; Jin, C.; Deng, T.G.; Sun, Y.; Li, X.; Hu, X.X.; Zhang, X.B.; et al. A multi-mitochondrial anticancer agent that selectively kills cancer cells and overcomes drug resistance. Chem. Med. Chem. 2017, 12, 250–256. [Google Scholar] [CrossRef] [Green Version]
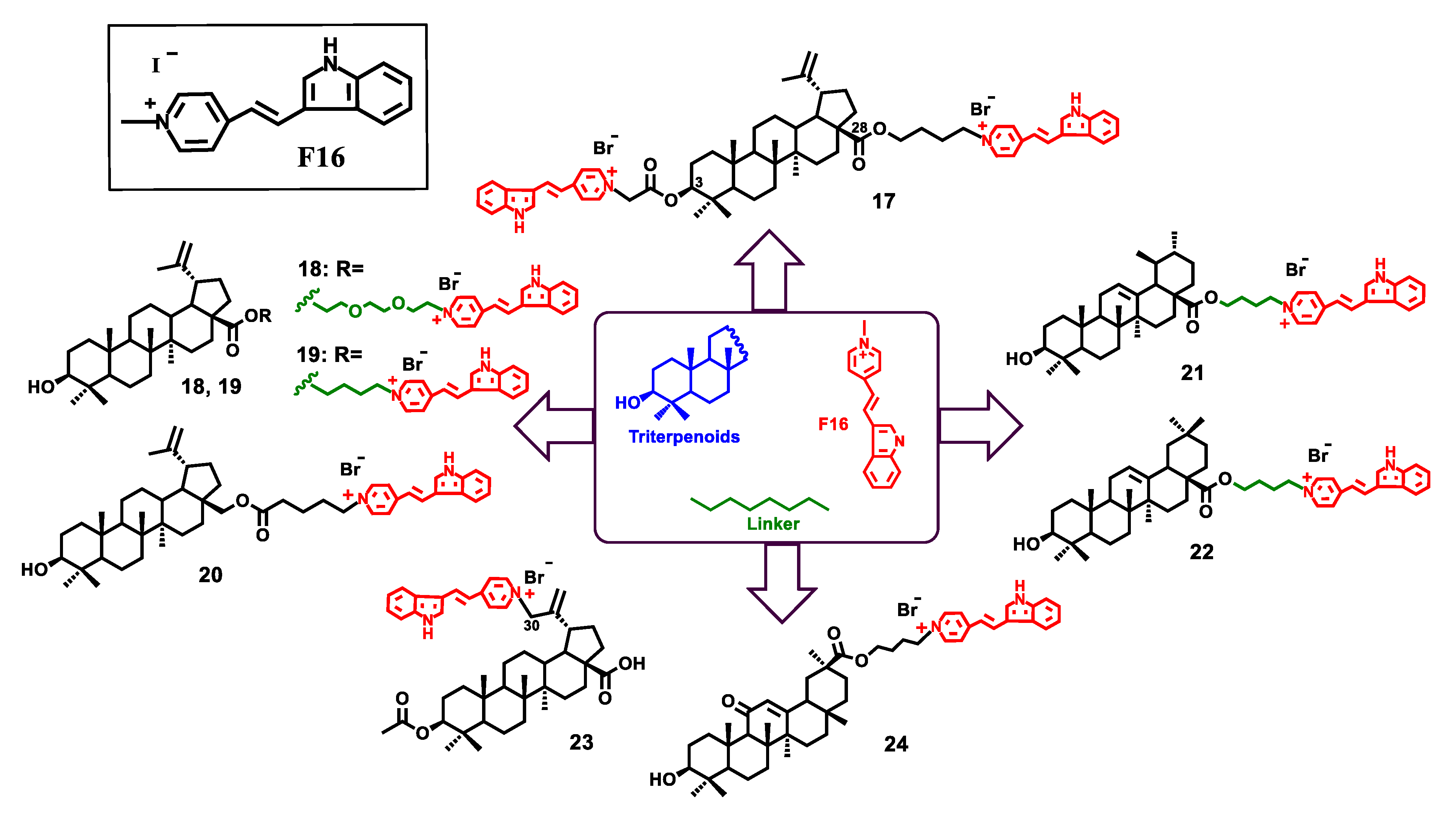
- Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Davletshin, E.V.; Tukhbatullin, A.A.; D’yakonov, V.A.; Yunusbaeva, M.M.; Dzhemileva, L.U.; Dzhemilev, U.M. Pentacyclic triterpene acid conjugated with mitochondria-targeting cation F16: Synthesis and evaluation of cytotoxic activities. Med. Chem. Res. 2021, 30, 940–951. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Semenova, A.A.; Ilzorkina, A.I.; Penkov, N.V.; Nedopekina, D.A.; Sharapov, V.A.; Khoroshavina, E.I.; Davletshin, E.V.; Belosludtseva, N.V.; Spivak, A.Y.; et al. Mitochondria-targeted prooxidant effects of betulinic acid conjugated with delocalized lipophilic cation F16. Free Radic Biol. Med. 2021, 168, 55–69. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Semenova, A.A.; Nedopekina, D.A.; Davletshin, E.V.; Spivak, A.Y.; Belosludtsev, K.N. Effect of F16-betulin conjugate on mitochondrial membranes and its role in cell death initiation. Membranes 2021, 11, 352. [Google Scholar] [CrossRef]
- Sommerwerk, S.; Heller, L.; Kerzig, C.; Kramell, A.E.; Csuk, R. Rhodamine B conjugates of triterpenoic acids are cytotoxic mitocans even at nanomolar concentrations. Eur. J. Med. Chem. 2017, 127, 1–9. [Google Scholar] [CrossRef]
- Csuk, R.; Siewert, B.; Dressel, C.; Schäfer, R. Tormentic acid derivatives: Synthesis and apoptotic activity. Eur. J. Med. Chem. 2012, 56, 237–245. [Google Scholar] [CrossRef]
- Wolfram, R.K.; Heller, L.; Csuk, R. Targeting mitochondria: Esters of rhodamine B with triterpenoids are mitocanic triggers of apoptosis. Eur. J. Med. Chem. 2018, 152, 21–30. [Google Scholar] [CrossRef]
- Wolfram, R.K.; Fischer, L.; Kluge, R.; Strohl, D.; Al-Harrasi, A.; Csuk, R. Homopiperazine-rhodamine B adducts of triterpenoic acids are strong mitocans. Eur. J. Med. Chem. 2018, 155, 869–879. [Google Scholar] [CrossRef]
- Kahnt, M.; Wiemann, J.; Fischer, L.; Sommerwerk, S.; Csuk, R. Transformation of asiatic acid into a mitocanic, bimodal-acting rhodamine B conjugate of nanomolar cytotoxicity. Eur. J. Med. Chem. 2018, 159, 143–148. [Google Scholar] [CrossRef]
- Serbian, I.; Hoenke, S.; Kraft, O.; Csuk, R. Ester and amide derivatives of rhodamine B exert cytotoxic effects on different human tumor cell lines. Med. Chem. Res. 2020, 29, 1655–1661. [Google Scholar] [CrossRef]
- Hoenke, S.; Serbian, I.; Deigner, H.-P.; Csuk, R. Mitocanic di- and triterpenoid rhodamine B conjugates. Molecules 2020, 25, 5443. [Google Scholar] [CrossRef]
- Serbian, I.; Hoenke, S.; Csuk, R. Synthesis of some steroidal mitocans of nanomolar cytotoxicity acting by apoptosis. Eur. J. Med. Chem. 2020, 199, 112425. [Google Scholar] [CrossRef]
- Macasoi, I.; Mioc, M.; Vaduva, D.B.; Ghiulai, R.; Mioc, A.; Soica, C.; Muntean, D.; Dumitrascu, V. In silico evaluation of the antiproliferative mitohocondrial targeted mechanism of action of some pentacyclic triterpene derivatives. Rev. Chim. 2019, 69, 3361–3363. [Google Scholar] [CrossRef]
- Panina, S.B.; Pei, J.; Kirienko, N.V. Mitochondrial metabolism as a target for acute myeloid leukemia treatment. Cancer Metab. 2021, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, L.X.; Grasso, D.; Bouzin, C.; Brusa, D.; Rossignol, R.; Sonveaux, P. Mitochondria participate in chemoresistance to cisplatin in human ovarian cancer cells. Mol. Cancer Res. 2020, 18, 1379–1391. [Google Scholar] [CrossRef]
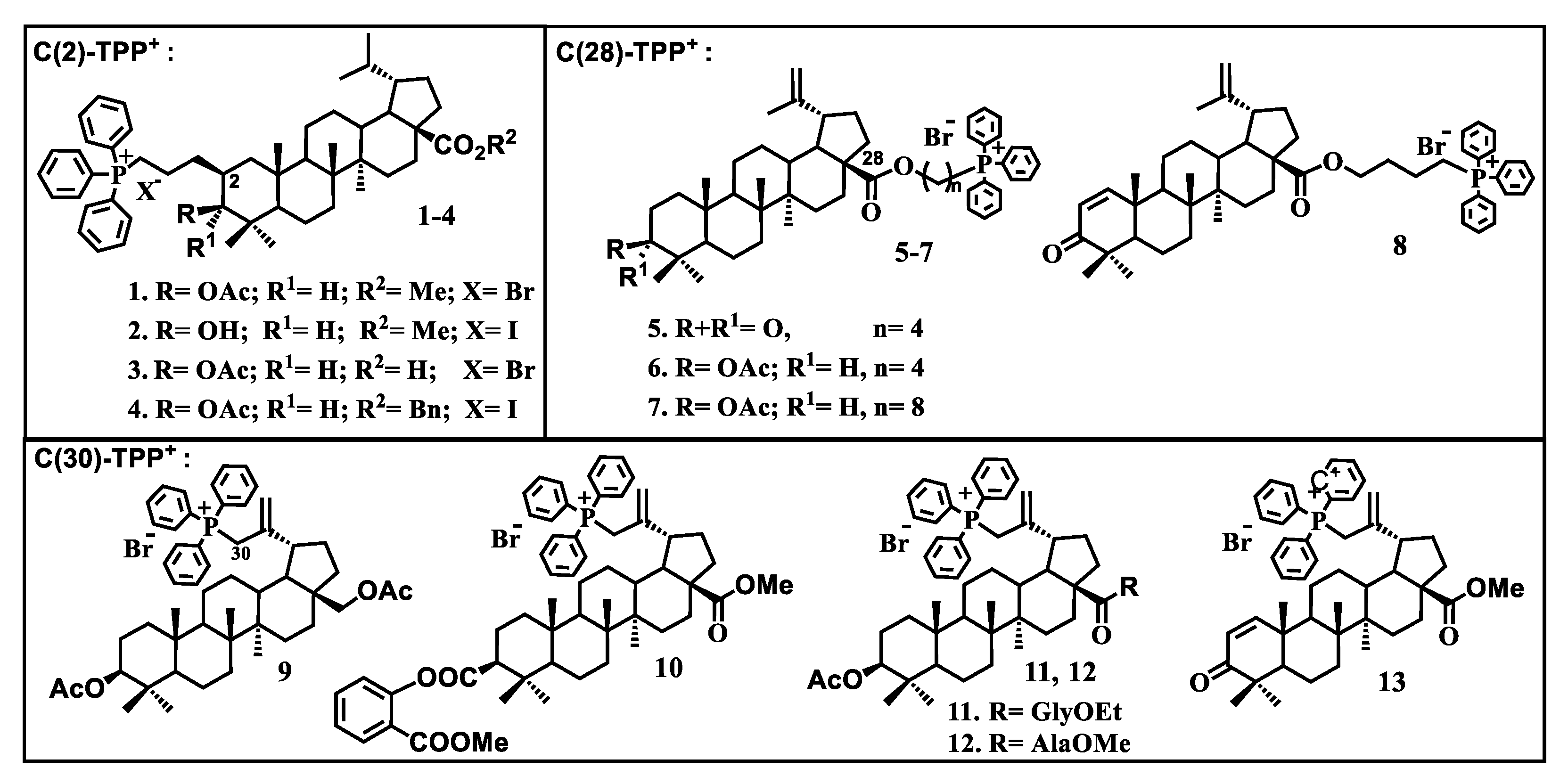
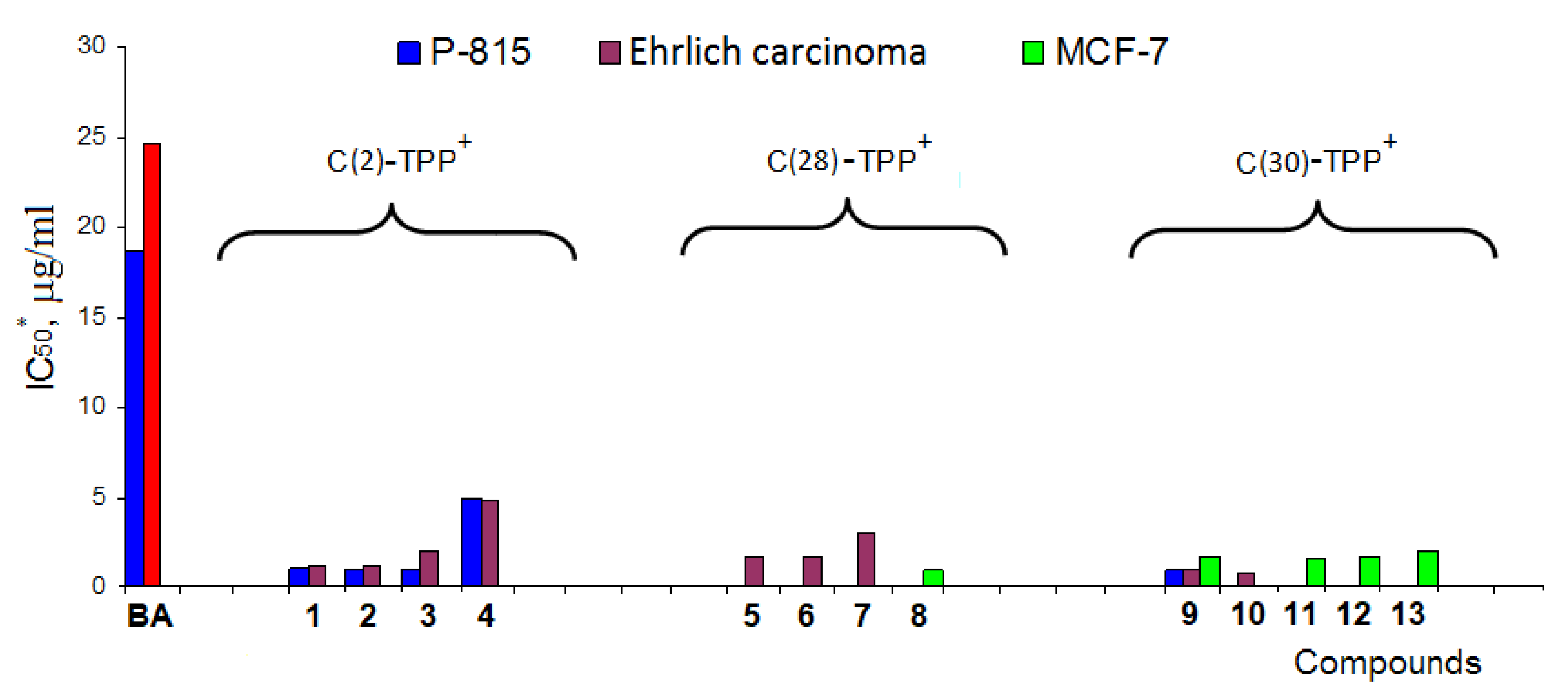

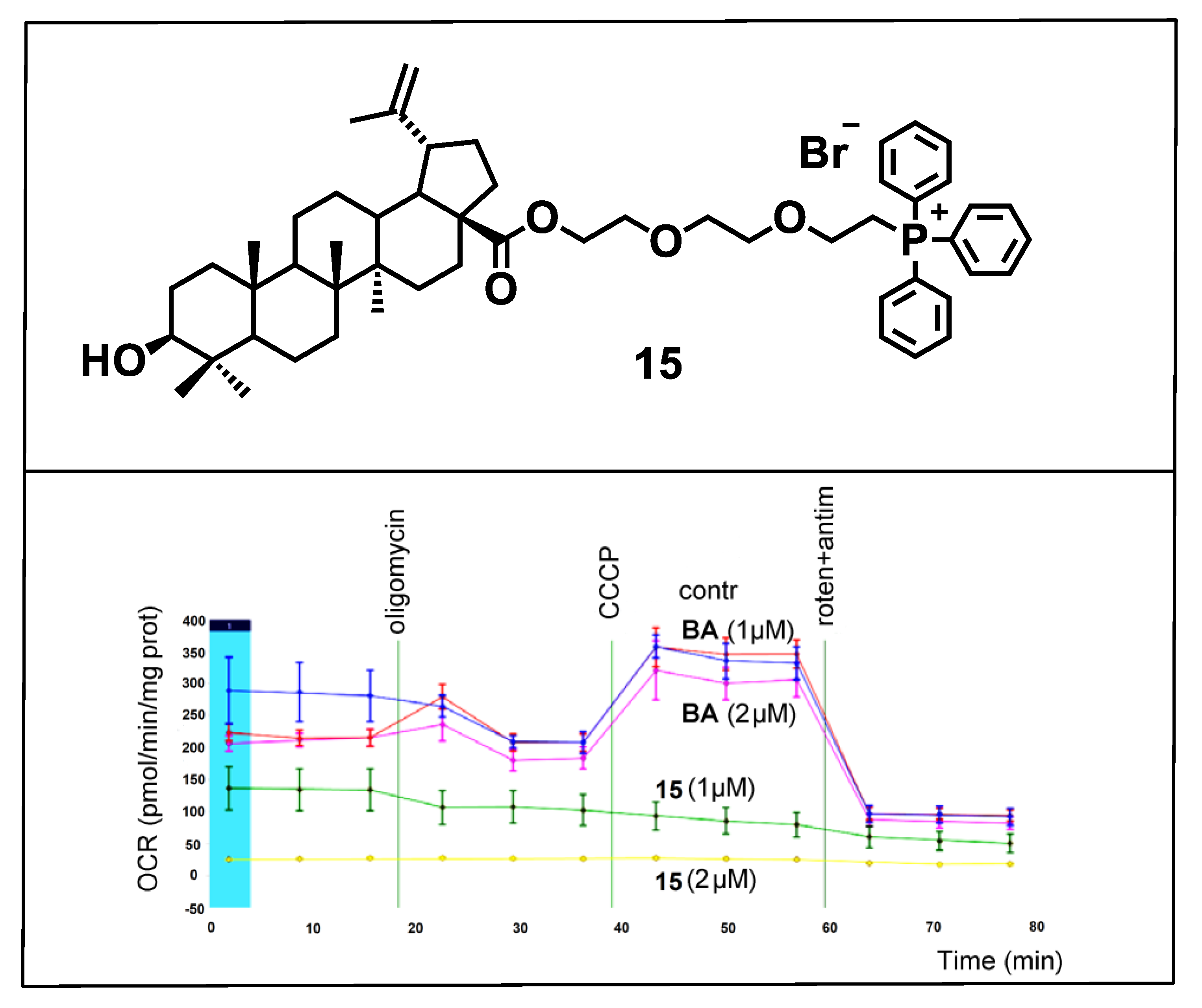
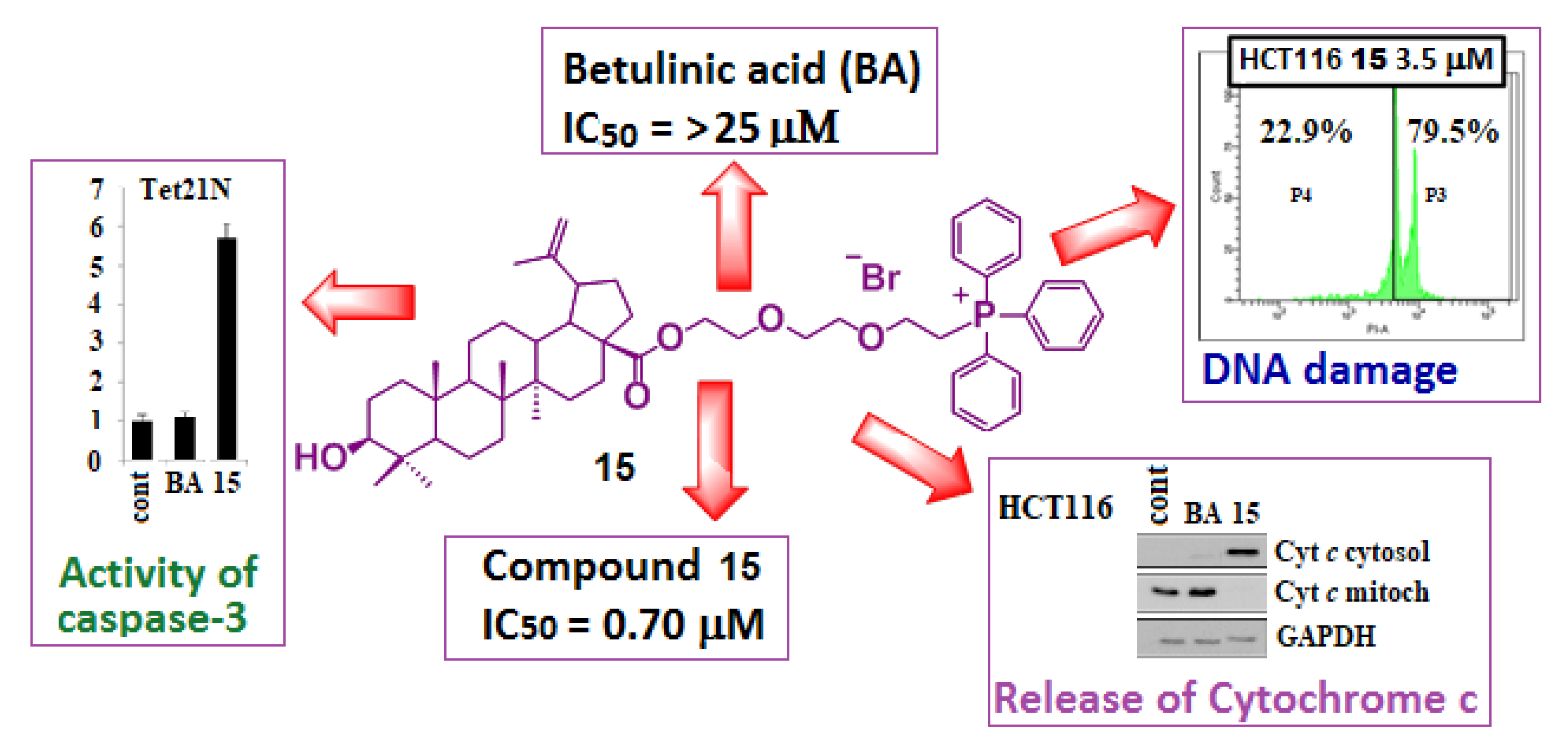



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
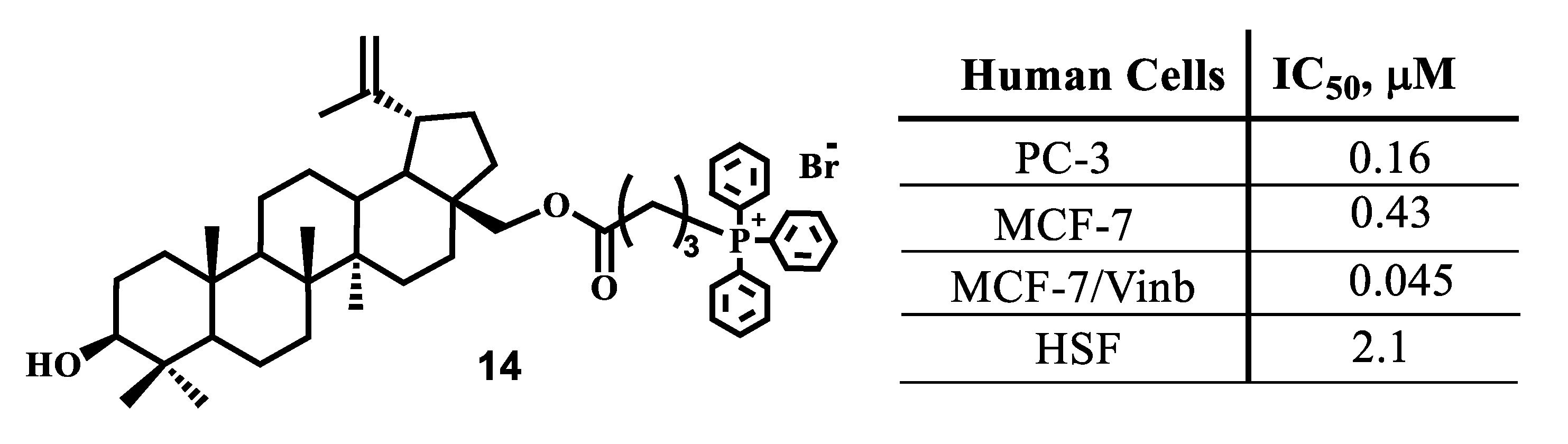{kind=link}
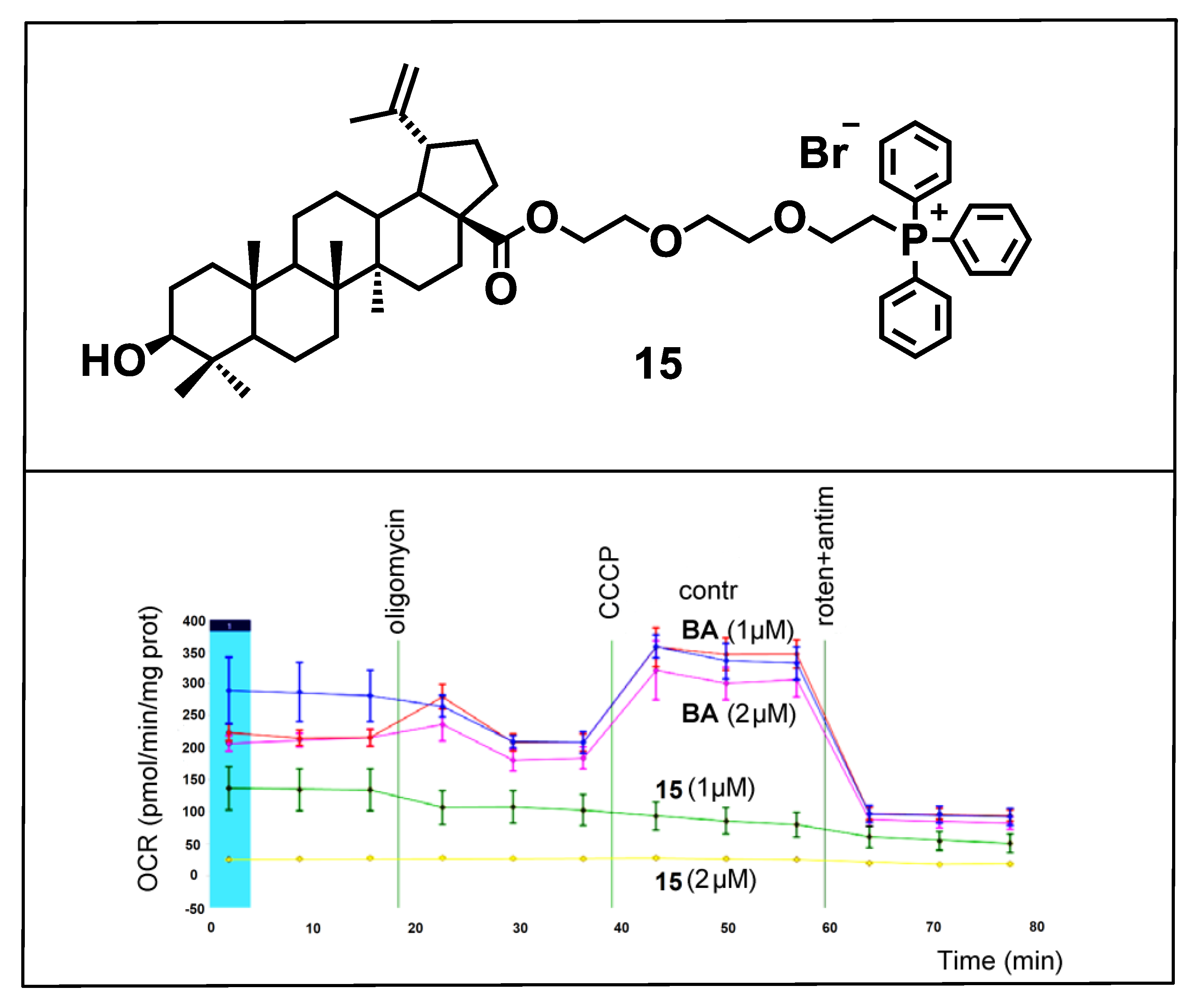{kind=link}
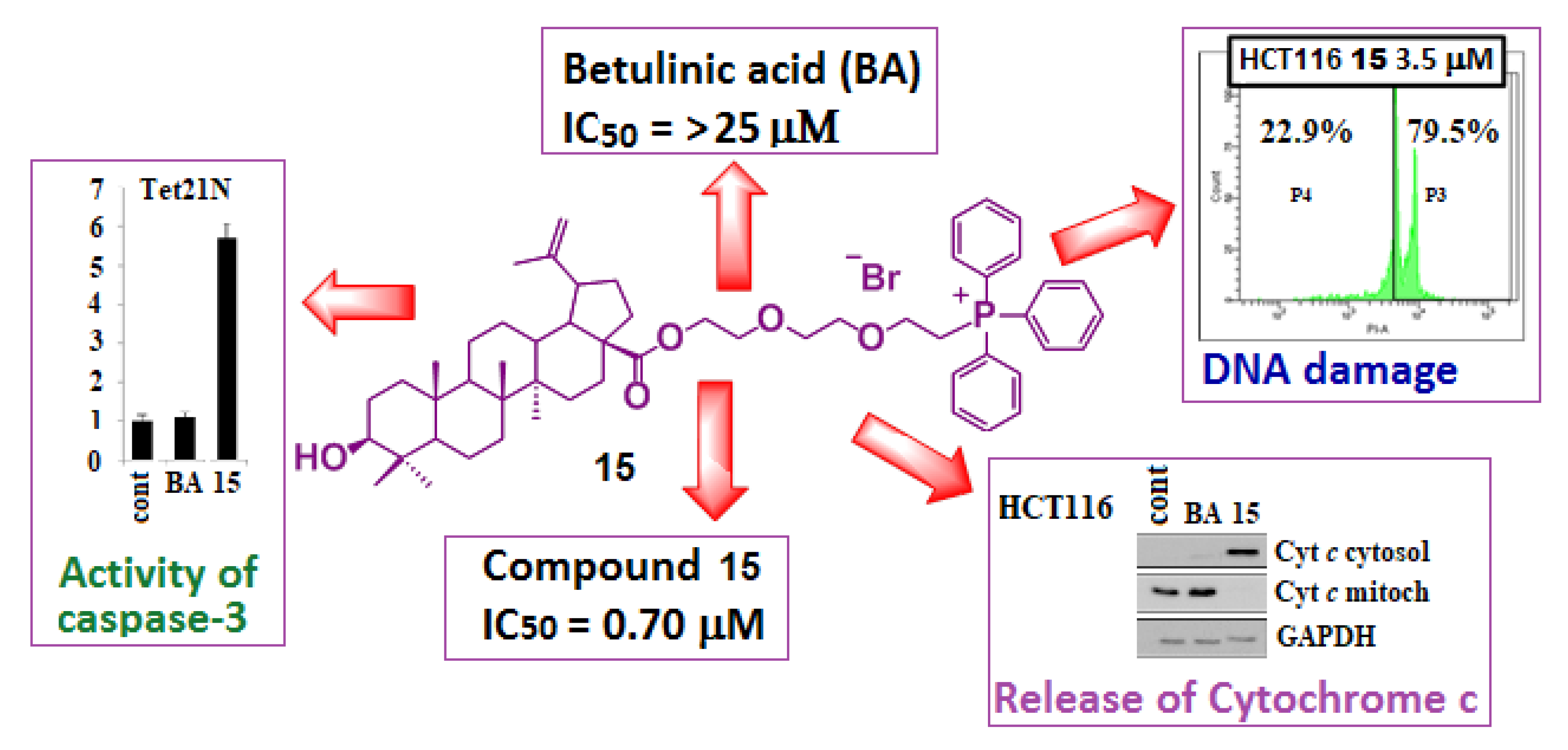{kind=link}
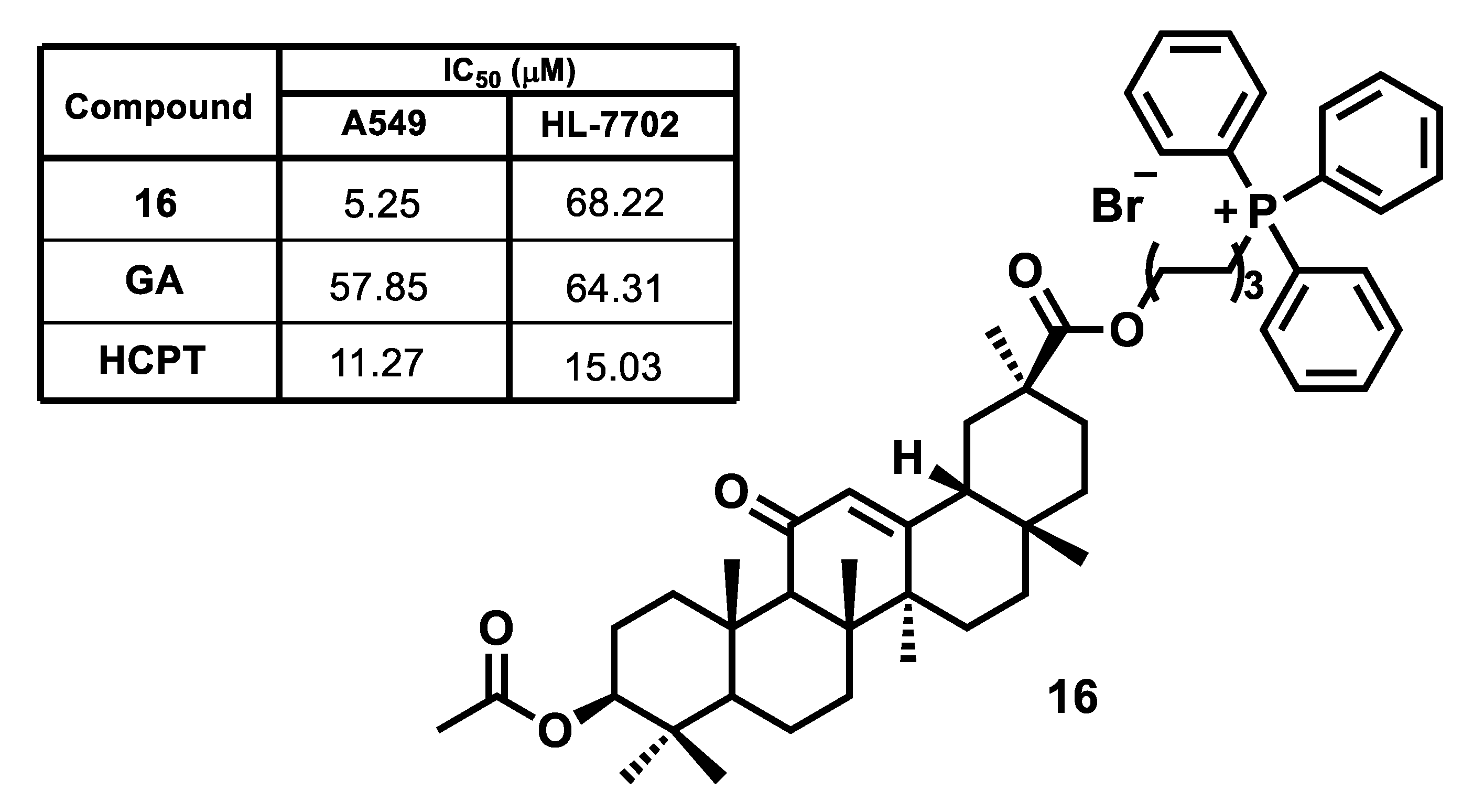{kind=link}
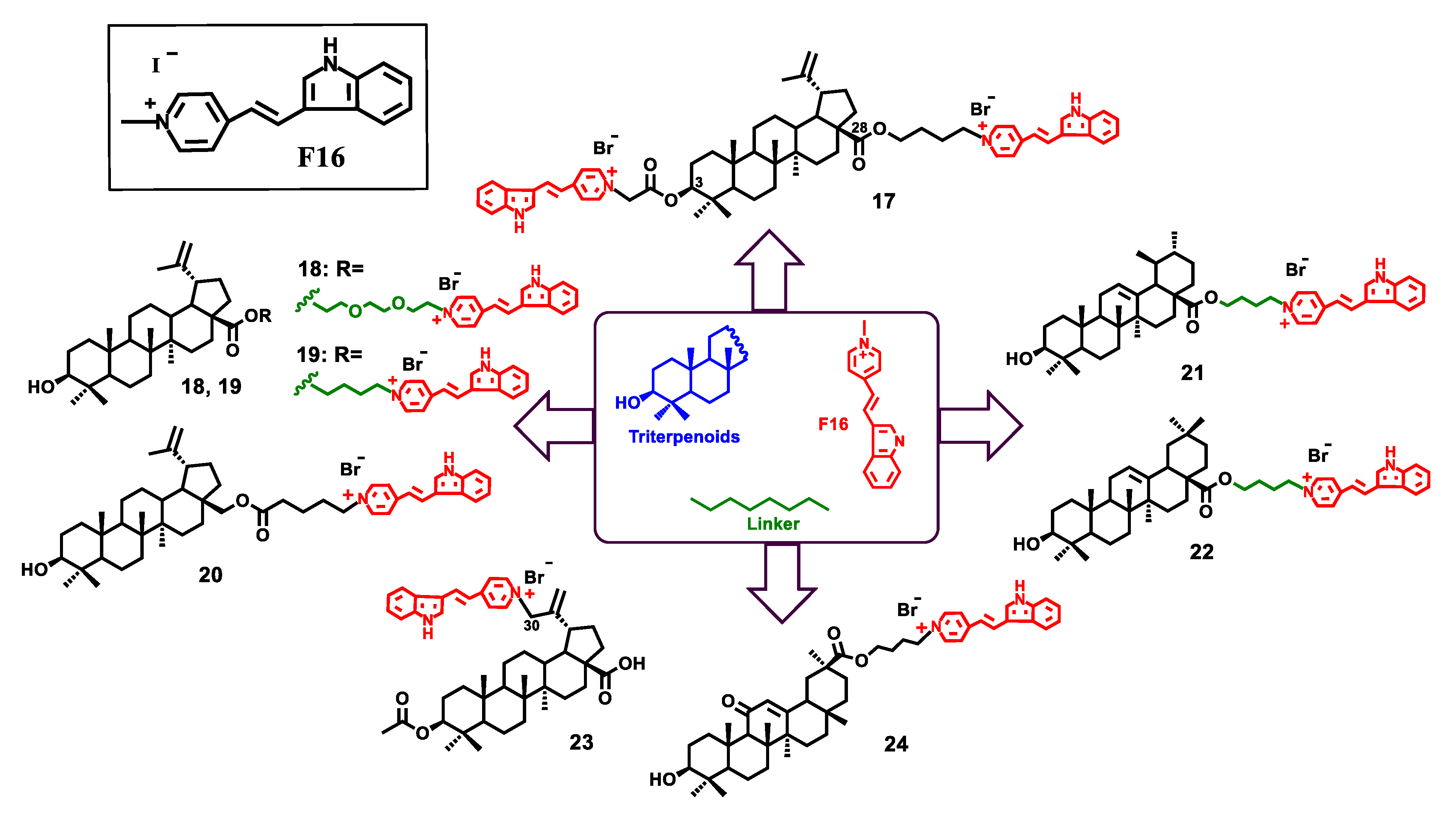{kind=link}
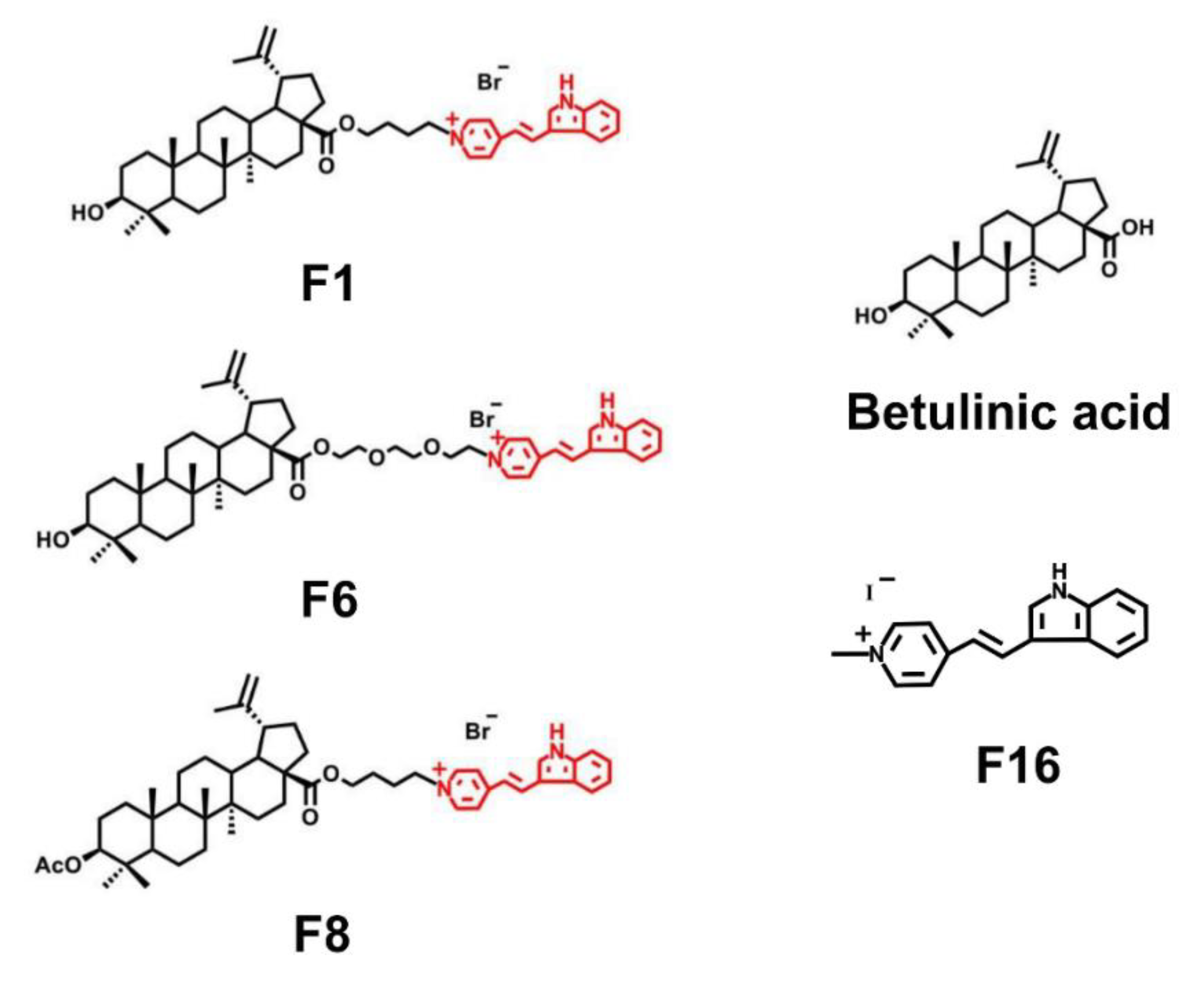{kind=link}
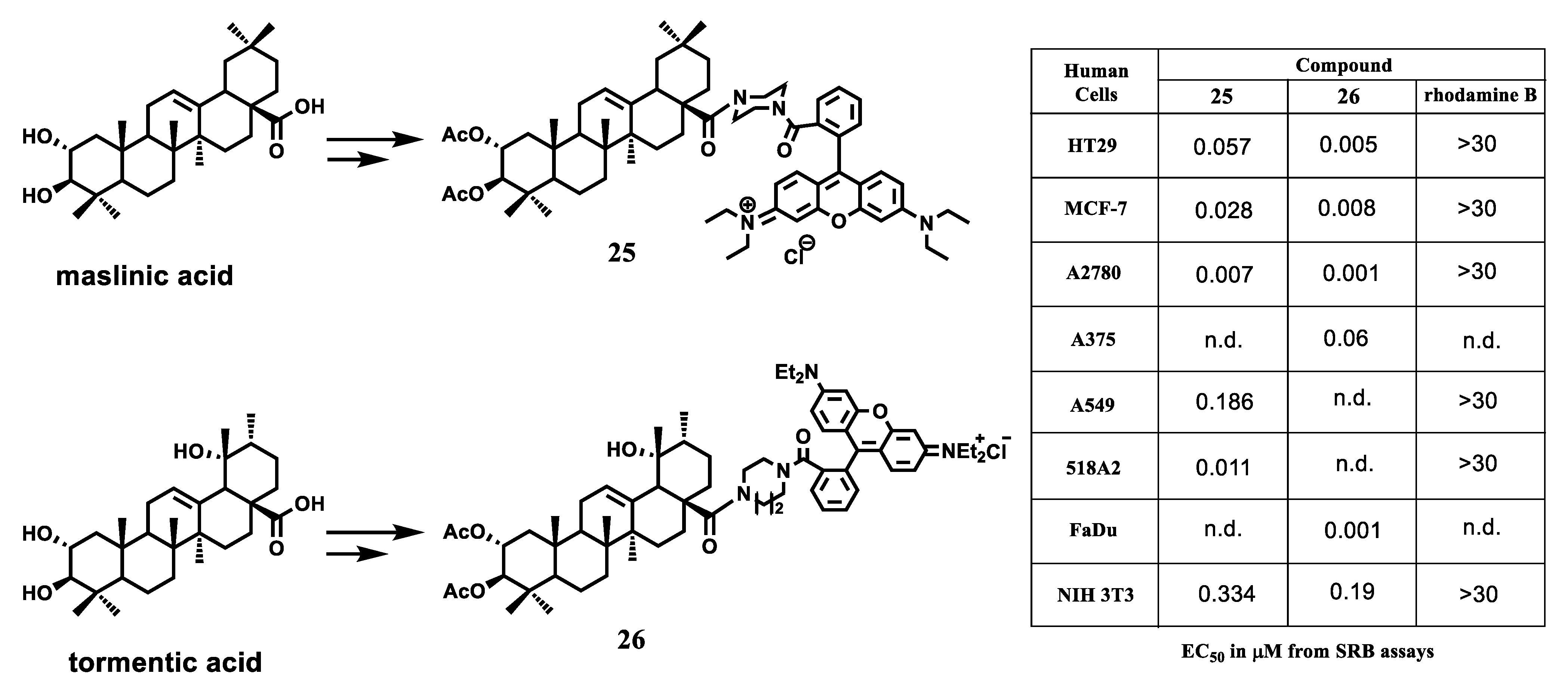{kind=link}
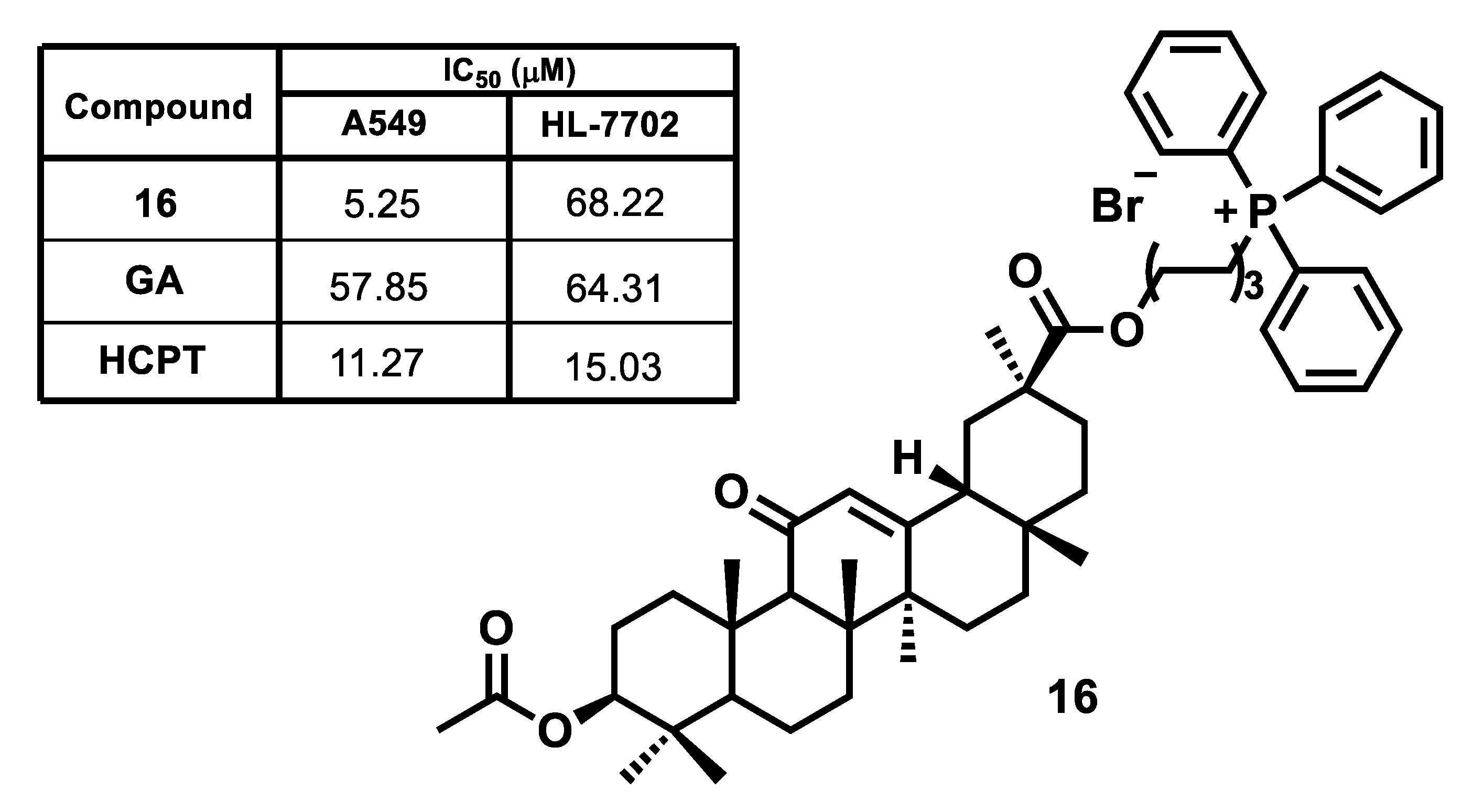
| Compound | IC50 (μM) a | |||
|---|---|---|---|---|
| U937 | Jurkat | K562 | Fibroblasts | |
| 17 | 4.190 ± 0.117 b | 4.360 ± 0.122 b | 4.010 ± 0.109 b | 10.400 ± 1.230 b |
| 18 | 0.573 ± 0.024 b | 1.260 ± 0.042 b | 1.210 ± 0.041 b | 5.500 ± 0.340 b |
| 19 | 0.616 ± 0.028 b | 0.844 ± 0.034 b | 0.812 ± 0.032 b | 6.100 ± 0.220 b |
| 20 | 0.906 ± 0.037 b | 0.937 ± 0.032 b | 0.904 ± 0.033 b | 8.200 ± 0.630 b |
| 21 | 2.461 ± 0.085 b | 0.623 ± 0.031 b | 0.588 ± 0.032 b | 6.230 ± 0.850 b |
| 22 | 0.607 ± 0.027 b | 0.687 ± 0.034 b | 0.671 ± 0.035 b | 3.490 ± 0.560 b |
| 23 | >125 b | >125 b | >125 b | >125 b |
| 24 | 2.425 ± 0.083 b | 0.559 ± 0.024 b | 0.511 ± 0.022 b | 8.300 ± 1.190 b |
| F16 | >500 b | >500 b | >500 b | >500 b |
| BAc | 149.290 ± 4.170 b | 81.680 ± 1.820 b | 78.540 ± 1.760 b | 236.400 ± 3.600 b |
| F16:BA/1:1 | 122.170 ± 3.460 b | 91.580 ± 1.950 b | 89.150 ± 1.890 b | 280.100 ± 3.440 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Dubinin, M.V.; Belosludtsev, K.N. Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: Selective Targeting Cancer Cell Mitochondria. J. Pers. Med. 2021, 11, 470. https://doi.org/10.3390/jpm11060470
Spivak AY, Nedopekina DA, Gubaidullin RR, Dubinin MV, Belosludtsev KN. Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: Selective Targeting Cancer Cell Mitochondria. Journal of Personalized Medicine. 2021; 11(6):470. https://doi.org/10.3390/jpm11060470
Chicago/Turabian StyleSpivak, Anna Yu., Darya A. Nedopekina, Rinat R. Gubaidullin, Mikhail V. Dubinin, and Konstantin N. Belosludtsev. 2021. "Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: Selective Targeting Cancer Cell Mitochondria" Journal of Personalized Medicine 11, no. 6: 470. https://doi.org/10.3390/jpm11060470
APA StyleSpivak, A. Y., Nedopekina, D. A., Gubaidullin, R. R., Dubinin, M. V., & Belosludtsev, K. N. (2021). Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: Selective Targeting Cancer Cell Mitochondria. Journal of Personalized Medicine, 11(6), 470. https://doi.org/10.3390/jpm11060470