
Identification of Novel Mutations in Colorectal Cancer Patients Using AmpliSeq Comprehensive Cancer Panel

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Description and Sample Collection
2.2. Ethical Approval
2.3. DNA Extraction
2.4. Comprehensive Cancer Panel (CCP) and Data Availability
2.5. Library Preparation and NGS Data Analysis
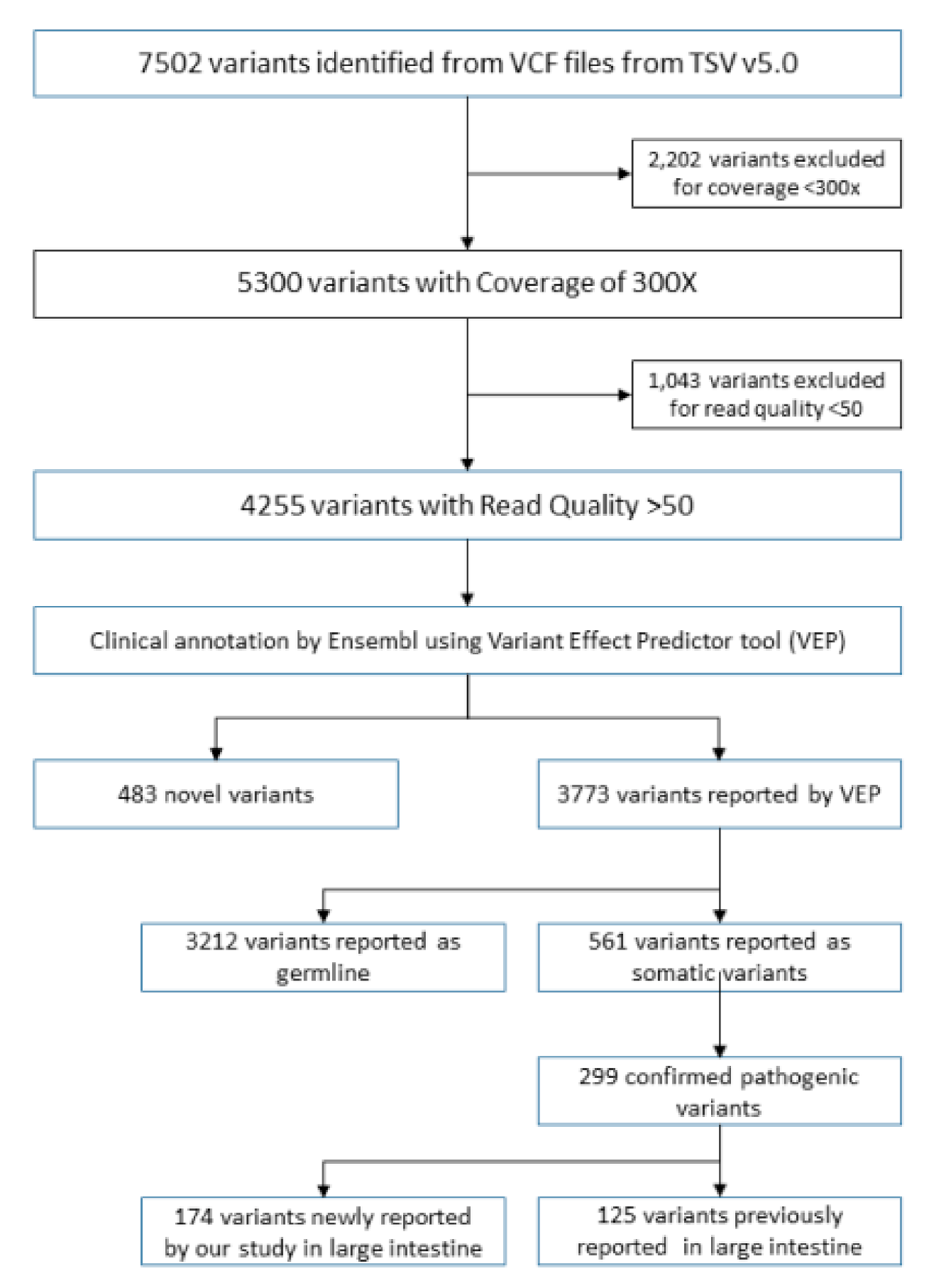
2.6. Variant Calling and Annotation
2.7. Molecular Profiling and Statistical Analysis
2.8. Ingenuity Pathway Analysis
3. Results
3.1. Cohort Characteristics
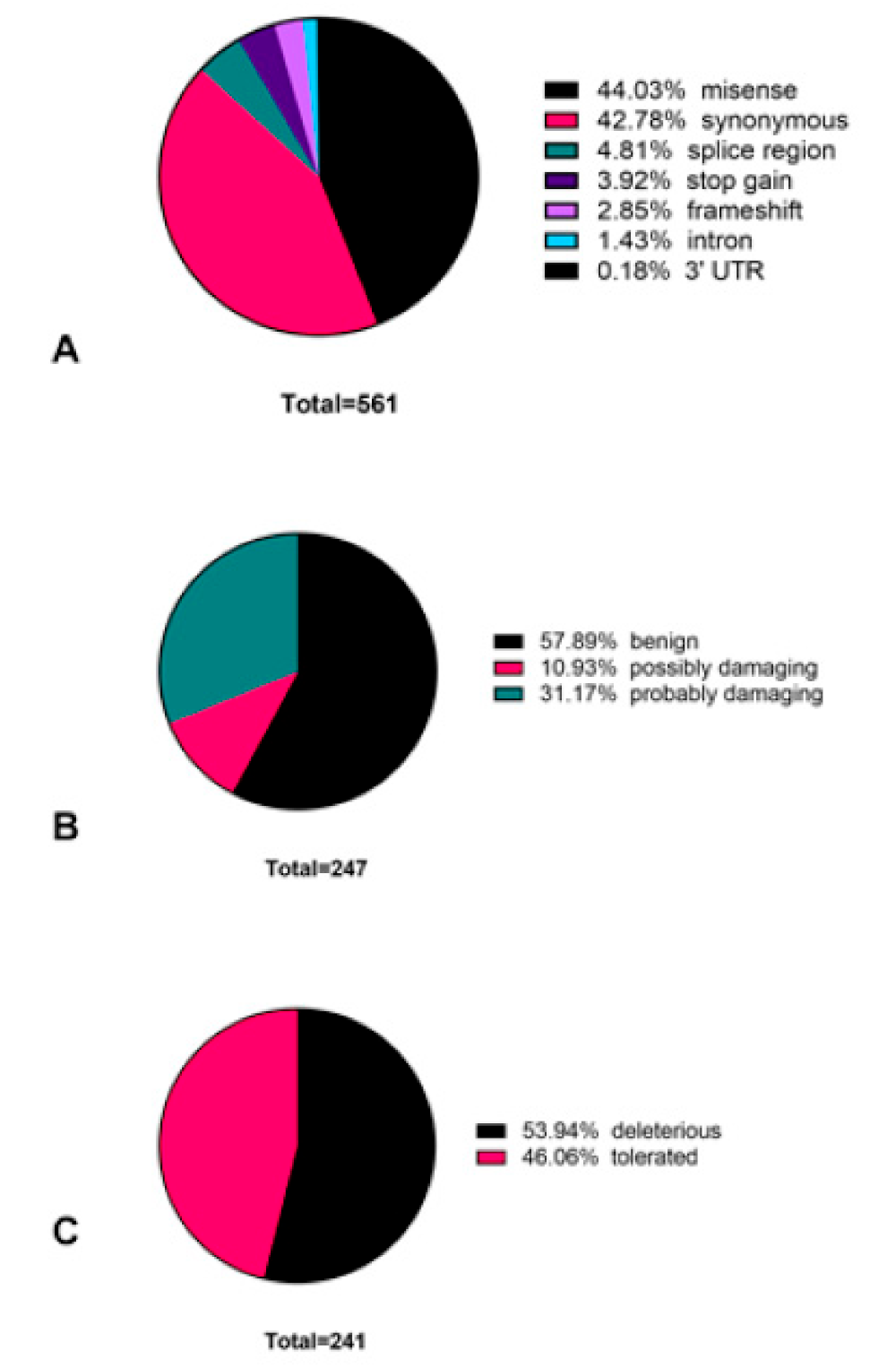
3.2. Novel Variants Identified in Colorectal Cancer Patient Cohort
3.3. Novel Variants Identified in Most Commonly Mutated APC, RET, and EGFR Genes
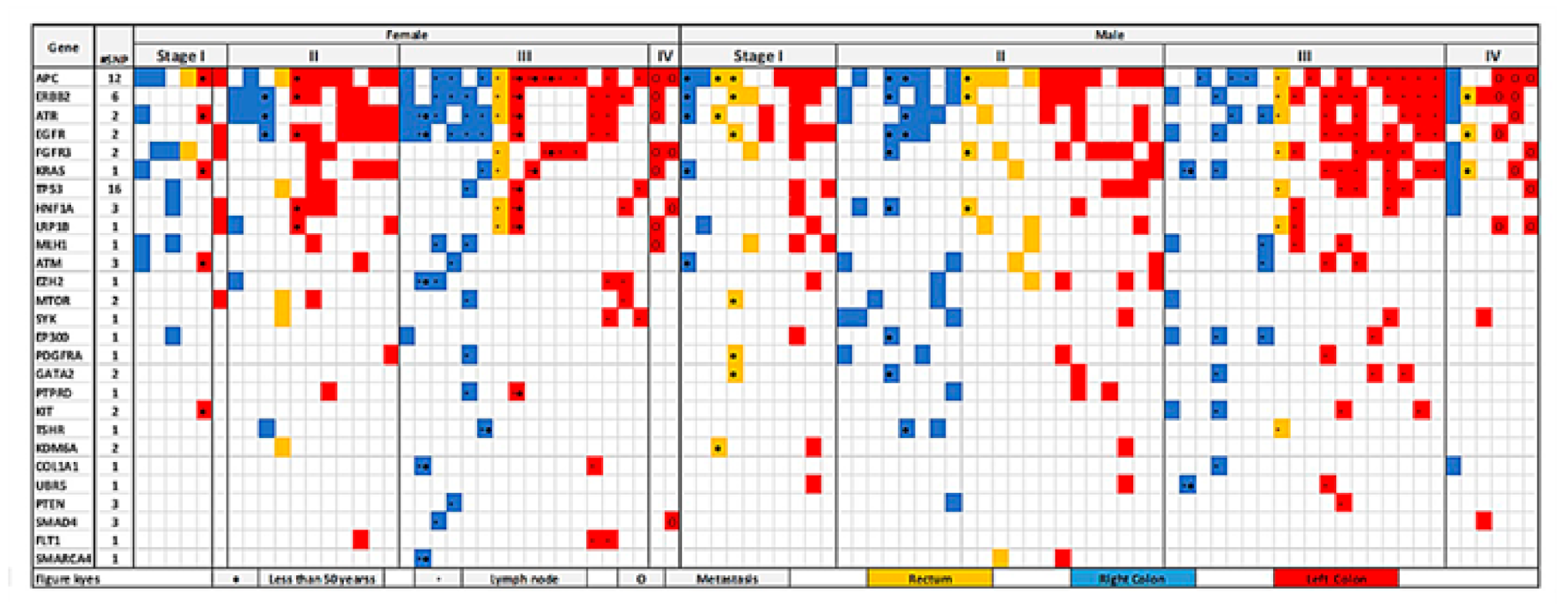
3.4. Colorectal-Cancer-Specific Variants Mapped to Twenty-Seven Genes
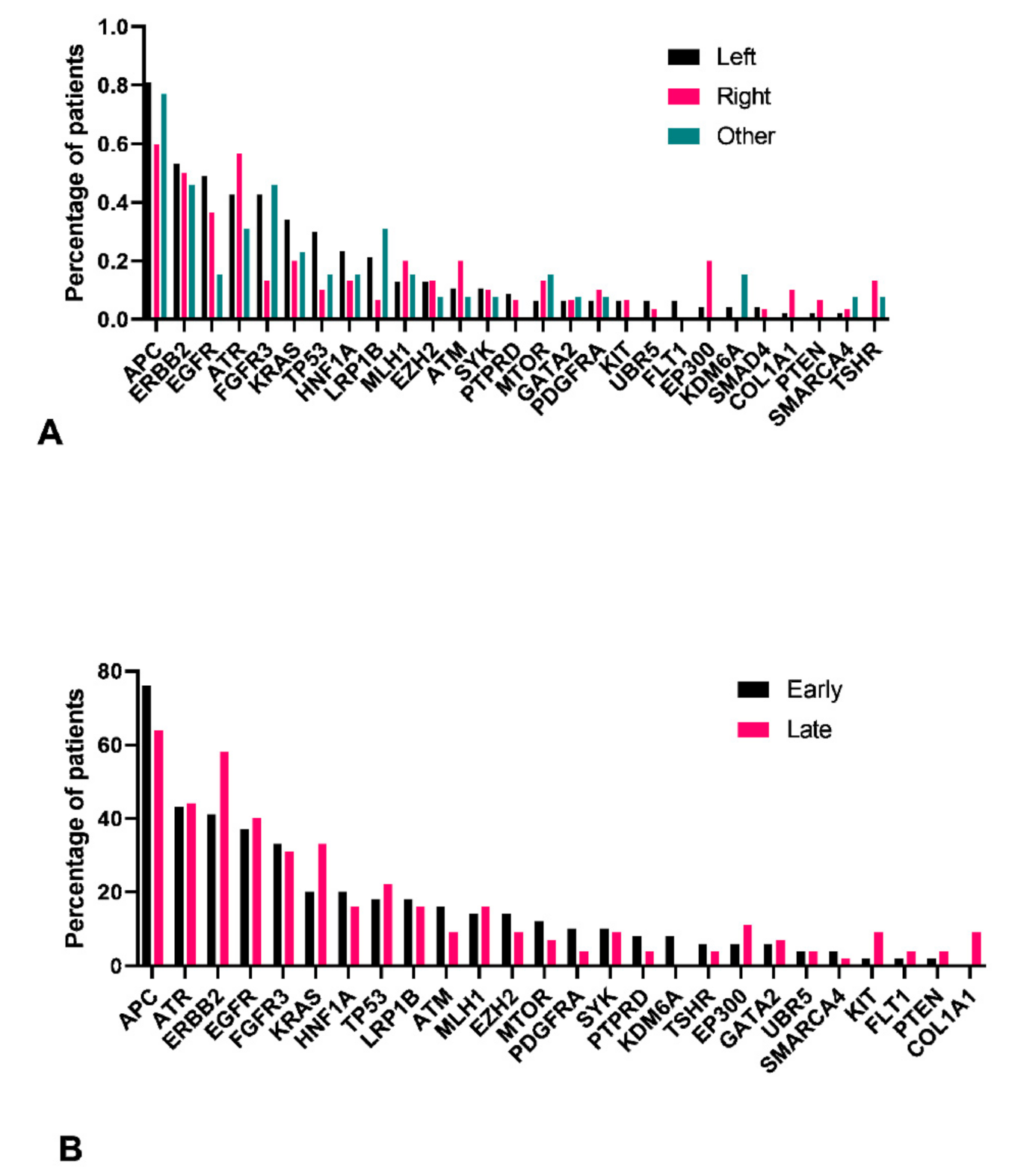
3.5. Left Colon Exhibits Higher Mutation Load
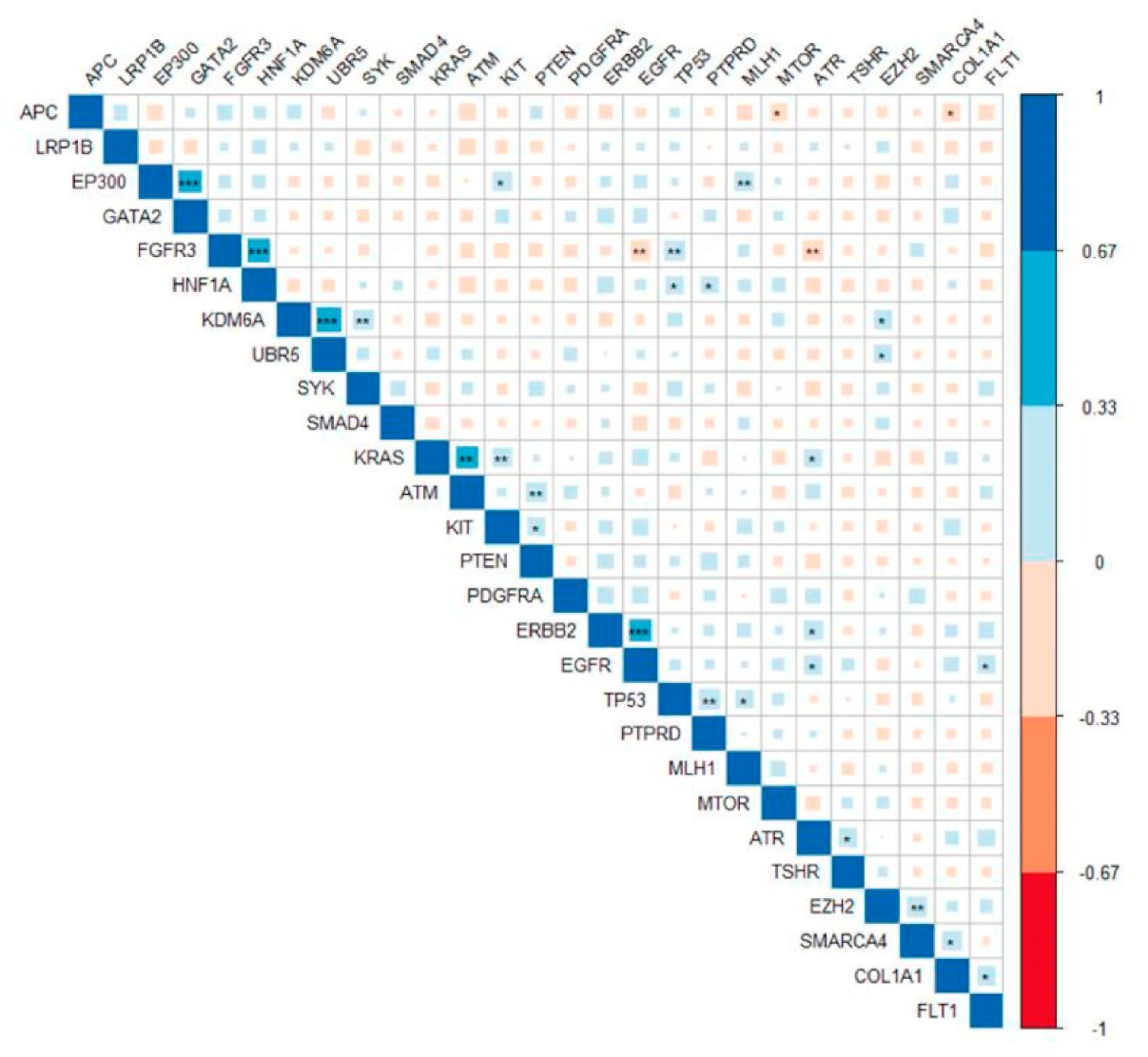
3.6. Pathogenic Variants in ERBB2 Were Significantly Correlated with Mutations in EGFR and ATR
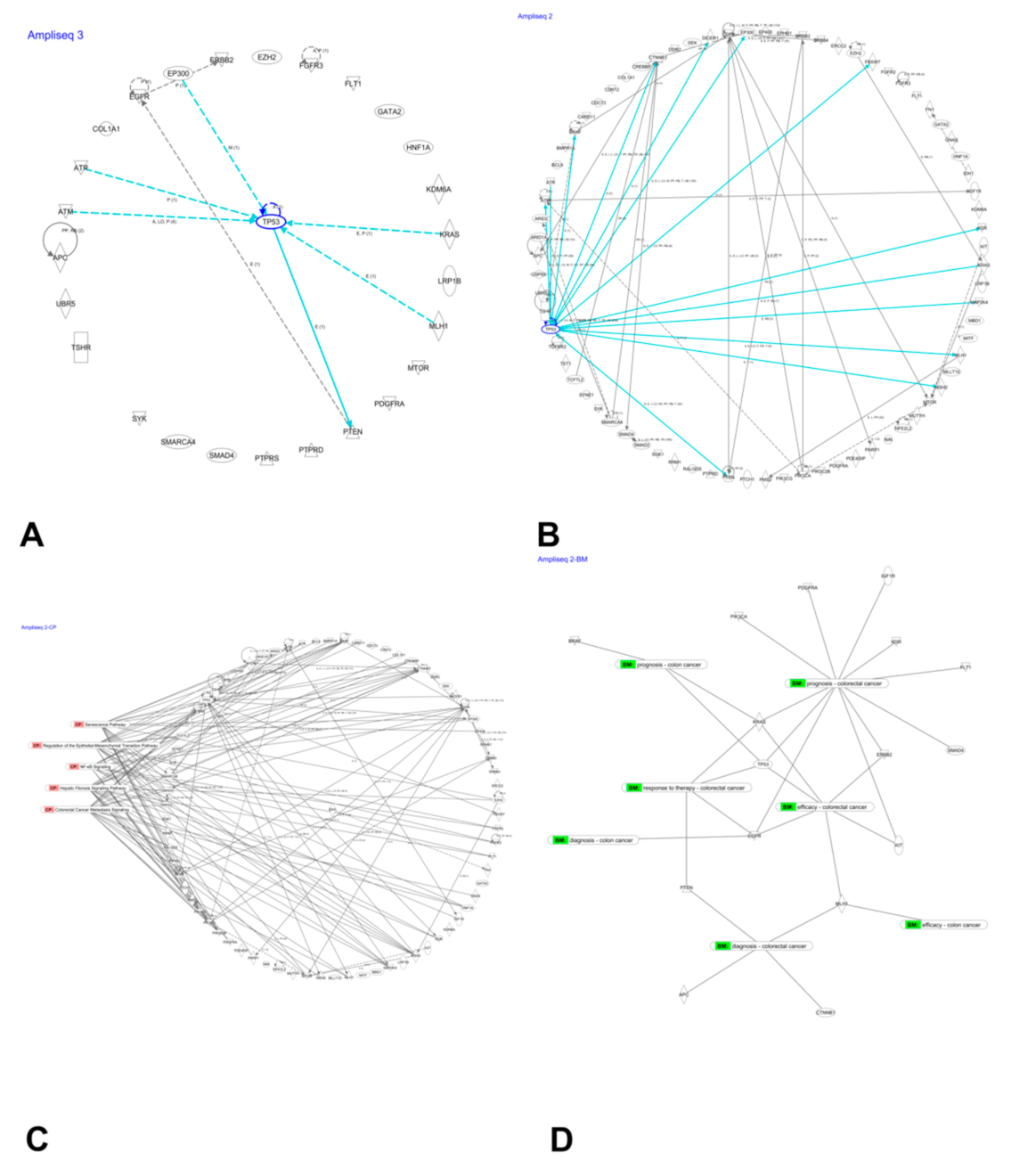
3.7. Network Analysis of Mutated Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rashid, M.; Vishwakarma, R.K.; Deeb, A.M.; Hussein, M.A.; Aziz, M.A. Molecular classification of colorectal cancer using the gene expression profile of tumor samples. Exp. Biol. Med. 2019, 244, 1005–1016. [Google Scholar] [CrossRef]
- Zhunussova, G.; Afonin, G.; Abdikerim, S.; Jumanov, A.; Perfilyeva, A.; Kaidarova, D.; Djansugurova, L. Mutation Spectrum of Cancer-Associated Genes in Patients With Early Onset of Colorectal Cancer. Front. Oncol. 2019, 9, 673. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, W.; Sobanski, T.; de Carvalho, A.C.; Evangelista, A.F.; Matsushita, M.; Berardinelli, G.N.; de Oliveira, M.A.; Reis, R.M.; Guimaraes, D.P. Mutation profiling of cancer drivers in Brazilian colorectal cancer. Sci. Rep. 2019, 9, 13687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alyabsi, M.; Alhumaid, A.; Allah-Bakhsh, H.; Alkelya, M.; Aziz, M.A. Colorectal cancer in Saudi Arabia as the proof-of-principle model for implementing strategies of predictive, preventive, and personalized medicine in healthcare. EPMA J. 2020, 11, 119–131. [Google Scholar] [CrossRef]
- Aziz, M.A.; Allah-Bakhsh, H. Colorectal cancer: A looming threat, opportunities, and challenges for the Saudi population and its healthcare system. Saudi J. Gastroenterol. 2018, 24, 196–197. [Google Scholar] [CrossRef]
- Aziz, M.A. Precision medicine in colorectal cancer. Saudi J. Gastroenterol. 2019, 25, 139–140. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Vitiello, P.P.; Marsoni, S.; Siena, S.; Tabernero, J.; Trusolino, L.; Bernards, R.; Bardelli, A. Precision oncology in metastatic colorectal cancer—From biology to medicine. Nat. Rev. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Sammarco, G.; Gallo, G.; Vescio, G.; Picciariello, A.; De Paola, G.; Trompetto, M.; Curro, G.; Ammendola, M. Mast Cells, microRNAs and Others: The Role of Translational Research on Colorectal Cancer in the Forthcoming Era of Precision Medicine. J. Clin. Med. 2020, 9, 2852. [Google Scholar] [CrossRef] [PubMed]
- Pellino, G.; Gallo, G.; Pallante, P.; Capasso, R.; De Stefano, A.; Maretto, I.; Malapelle, U.; Qiu, S.; Nikolaou, S.; Barina, A.; et al. Noninvasive Biomarkers of Colorectal Cancer: Role in Diagnosis and Personalised Treatment Perspectives. Gastroenterol. Res. Pract. 2018, 2018, 2397863. [Google Scholar] [CrossRef] [PubMed]
- Dallol, A.; Buhmeida, A.; Al-Ahwal, M.S.; Al-Maghrabi, J.; Bajouh, O.; Al-Khayyat, S.; Alam, R.; Abusanad, A.; Turki, R.; Elaimi, A.; et al. Clinical significance of frequent somatic mutations detected by high-throughput targeted sequencing in archived colorectal cancer samples. J. Transl. Med. 2016, 14, 118. [Google Scholar] [CrossRef] [Green Version]
- Torrent Suite Version 5.8; Thermo Fisher Scientific: South San Francisco, CA, USA, 2018.
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Emond, M.J.; Bamshad, M.J.; Barnes, K.C.; Rieder, M.J.; Nickerson, D.A.; Christiani, D.C.; Wurfel, M.M.; Lin, X. Optimal Unified Approach for Rare-Variant Association Testing with Application to Small-Sample Case-Control Whole-Exome Sequencing Studies. Am. J. Hum. Genet. 2012, 91, 224–237. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Paraskeva, C.; Markowitz, S.; Willson, J.K.; Hamilton, S.; Vogelstein, B. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990, 50, 7717–7722. [Google Scholar]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Huang, J.; Papadopoulos, N.; McKinley, A.J.; Farrington, S.M.; Curtis, L.J.; Wyllie, A.H.; Zheng, S.; Willson, J.K.; Markowitz, S.D.; Morin, P.; et al. APC mutations in colorectal tumors with mismatch repair deficiency. Proc. Natl. Acad. Sci. USA 1996, 93, 9049–9054. [Google Scholar] [CrossRef] [Green Version]
- Jen, J.; Kim, H.; Piantadosi, S.; Liu, Z.F.; Levitt, R.C.; Sistonen, P.; Kinzler, K.W.; Vogelstein, B.; Hamilton, S.R. Allelic loss of chromosome 18q and prognosis in colorectal cancer. N. Engl. J. Med. 1994, 331, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Apoptosis and APC in colorectal tumorigenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 7950–7954. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.A.; Yousef, Z.; Saleh, A.M.; Mohammad, S.; Al Knawy, B. Towards personalized medicine of colorectal cancer. Crit. Rev. Oncol. Hematol. 2017, 118, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Sharma, Y.; Miladi, M.; Dukare, S.; Boulay, K.; Caudron-Herger, M.; Gross, M.; Backofen, R.; Diederichs, S. A pan-cancer analysis of synonymous mutations. Nat. Commun. 2019, 10, 2569. [Google Scholar] [CrossRef] [Green Version]
- Foo, J.; Liu, L.L.; Leder, K.; Riester, M.; Iwasa, Y.; Lengauer, C.; Michor, F. An Evolutionary Approach for Identifying Driver Mutations in Colorectal Cancer. PLoS Comput. Biol. 2015, 11, e1004350. [Google Scholar] [CrossRef]
- Huang, D.; Sun, W.; Zhou, Y.; Li, P.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190.e1173. [Google Scholar] [CrossRef] [Green Version]
- Aghagolzadeh, P.; Radpour, R. New trends in molecular and cellular biomarker discovery for colorectal cancer. World J. Gastroenterol. 2016, 22, 5678–5693. [Google Scholar] [CrossRef] [PubMed]
- Jauhri, M.; Bhatnagar, A.; Gupta, S.; Shokeen, Y.; Minhas, S.; Aggarwal, S. Targeted molecular profiling of rare genetic alterations in colorectal cancer using next-generation sequencing. Med. Oncol. 2016, 33, 106. [Google Scholar] [CrossRef]
- Song, H.N.; Lee, C.; Kim, S.T.; Kim, S.Y.; Kim, N.K.; Jang, J.; Kang, M.; Jang, H.; Ahn, S.; Kim, S.H.; et al. Molecular characterization of colorectal cancer patients and concomitant patient-derived tumor cell establishment. Oncotarget 2016, 7, 19610–19619. [Google Scholar] [CrossRef]
- Song, E.K.; Tai, W.M.; Messersmith, W.A.; Bagby, S.; Purkey, A.; Quackenbush, K.S.; Pitts, T.M.; Wang, G.; Blatchford, P.; Yahn, R.; et al. Potent antitumor activity of cabozantinib, a c-MET and VEGFR2 inhibitor, in a colorectal cancer patient-derived tumor explant model. Int. J. Cancer 2015, 136, 1967–1975. [Google Scholar] [CrossRef] [Green Version]
- Baran, B.; Mert Ozupek, N.; Yerli Tetik, N.; Acar, E.; Bekcioglu, O.; Baskin, Y. Difference Between Left-Sided and Right-Sided Colorectal Cancer: A Focused Review of Literature. Gastroenterol. Res. 2018, 11, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Missiaglia, E.; Jacobs, B.; D’Ario, G.; Di Narzo, A.F.; Soneson, C.; Budinska, E.; Popovici, V.; Vecchione, L.; Gerster, S.; Yan, P.; et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 2014, 25, 1995–2001. [Google Scholar] [CrossRef]
- LaPointe, L.C.; Dunne, R.; Brown, G.S.; Worthley, D.L.; Molloy, P.L.; Wattchow, D.; Young, G.P. Map of differential transcript expression in the normal human large intestine. Physiol. Genomics 2008, 33, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Young, G.P.; Rabeneck, L.; Winawer, S.J. The Global Paradigm Shift in Screening for Colorectal Cancer. Gastroenterology 2019, 156, 843–851.e842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, S.; Hou, Y.; Zhou, X.; Li, X.; Yong, J.; Wang, Y.; Wang, W.; Yan, J.; Hu, B.; Guo, H.; et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 2018, 362, 1060–1063. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Yang, Y.; Li, X.; Huang, M.; Xu, F.; Ge, W.; Zhang, S.; Zheng, S. Multi-omics Approach Reveals Distinct Differences in Left- and Right-Sided Colon Cancer. Mol. Cancer Res. 2018, 16, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Jia, P.; Zhao, Z. Impacts of somatic mutations on gene expression: An association perspective. Brief Bioinform. 2017, 18, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Molinari, C.; Marisi, G.; Passardi, A.; Matteucci, L.; De Maio, G.; Ulivi, P. Heterogeneity in Colorectal Cancer: A Challenge for Personalized Medicine? Int. J. Mol. Sci. 2018, 19, 3733. [Google Scholar] [CrossRef] [Green Version]
- Grizzle, W.E.; Bell, W.C.; Sexton, K.C. Issues in collecting, processing and storing human tissues and associated information to support biomedical research. Cancer Biomark 2010, 9, 531–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiltemann, S.; Jenster, G.; Trapman, J.; van der Spek, P.; Stubbs, A. Discriminating somatic and germline mutations in tumor DNA samples without matching normals. Genome Res. 2015, 25, 1382–1390. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; White, T.A.; MacKenzie, A.P.; Clegg, N.; Lee, C.; Dumpit, R.F.; Coleman, I.; Ng, S.B.; Salipante, S.J.; Rieder, M.J.; et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 17087–17092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.M.; Li, Q.; Yan, M.; Cao, H.; Hu, Y.; He, K.Y.; Cao, K.; Li, M.M.; Wang, K. Variant Interpretation for Cancer (VIC): A computational tool for assessing clinical impacts of somatic variants. Genome Med. 2019, 11, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age, Years (SD) | 62 (14) |
| Male, n (%) | 58 (61%) |
| Stage | |
| I, n (%) | 17 (18%) |
| II, n (%) | 32 (34%) |
| III, n (%) | 37 (39%) |
| IV, n (%) | 8 (9%) |
| Primary Tumor | |
| T1, n (%) | 2 (2%) |
| T2, n (%) | 18 (19%) |
| T3, n (%) | 61 (65%) |
| T4, n (%) | 13 (14%) |
| Lymph Node | |
| N0, n (%) | 55 (59%) |
| N1, n (%) | 33 (35%) |
| N2, n (%) | 6 (6%) |
| Distant Metastasis | |
| M0, n (%) | 90 (96%) |
| M1, n (%) | 4 (4%) |
| Site | |
| Left colon, n (%) | 47 (52%) |
| Right colon, n (%) | 30 (33%) |
| Rectum, n (%) | 13 (14%) |
| Description of Variants Variants and Individuals for the Top 20 Genes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Variants | Individual a | ||||||||
| Total | Novel | Pathogenic | Somatic | PolyPhen Damaging | SIFT Deleterious | Non-Synonymous | Pathogenic % | Somatic % | ||
| All Tissue | Tissue-Specific | |||||||||
| APC | 38 | 5 | 15 | 12 | 23 | 1 | 2 | 30 | 0.96 | 0.99 |
| RET | 13 | 0 | 2 | 0 | 3 | 3 | 3 | 6 | 0.53 | 0.83 |
| EGFR | 17 | 0 | 4 | 2 | 6 | 0 | 0 | 7 | 0.53 | 0.69 |
| LRP1B | 70 | 12 | 10 | 1 | 22 | 3 | 1 | 48 | 0.52 | 0.86 |
| ERBB2 | 14 | 3 | 3 | 3 | 4 | 1 | 2 | 10 | 0.51 | 0.52 |
| ATR | 31 | 6 | 4 | 2 | 11 | 1 | 0 | 19 | 0.46 | 0.68 |
| CSMD3 | 42 | 5 | 4 | 0 | 8 | 5 | 3 | 34 | 0.44 | 0.52 |
| RALGDS | 13 | 1 | 2 | 1 | 3 | 1 | 2 | 6 | 0.36 | 0.42 |
| HIF1A | 8 | 0 | 3 | 0 | 3 | 0 | 0 | 6 | 0.36 | 0.36 |
| FGFR3 | 18 | 2 | 2 | 2 | 3 | 1 | 0 | 11 | 0.33 | 0.34 |
| KRAS | 7 | 1 | 1 | 1 | 2 | 0 | 1 | 5 | 0.28 | 0.28 |
| PIK3CG | 14 | 1 | 4 | 1 | 5 | 1 | 4 | 7 | 0.22 | 0.23 |
| TP53 | 24 | 1 | 16 | 16 | 18 | 1 | 9 | 23 | 0.21 | 0.31 |
| HNF1A | 13 | 0 | 5 | 3 | 4 | 0 | 0 | 9 | 0.20 | 0.27 |
| PIK3R1 | 10 | 2 | 2 | 0 | 3 | 0 | 0 | 6 | 0.19 | 0.30 |
| KDM6A | 11 | 1 | 3 | 2 | 3 | 1 | 0 | 8 | 0.19 | 0.19 |
| ATM | 31 | 3 | 6 | 3 | 3 | 0 | 4 | 26 | 0.17 | 0.16 |
| MLH1 | 9 | 1 | 2 | 1 | 3 | 0 | 1 | 8 | 0.16 | 0.17 |
| PRDM1 | 5 | 0 | 2 | 0 | 2 | 0 | 1 | 3 | 0.16 | 0.16 |
| JAK1 | 11 | 0 | 3 | 0 | 7 | 0 | 0 | 3 | 0.14 | 0.44 |
| Top genes associated with female versus male (232). | SetID | p.Value | N.Marker.All | N.Marker.Test | MAC | m | Method.bin | MAP |
| AR | 0.0002 | 5 | 5 | 37 | 19 | QA | −1 | |
| BTK | 0.003 | 1 | 1 | 5 | 5 | ER | 0.003523 | |
| SAMD9 | 0.003 | 14 | 14 | 66 | 30 | QA | −1 | |
| PAX7 | 0.006605 | 6 | 6 | 41 | 26 | QA | −1 | |
| KDM6A | 0.010983 | 11 | 11 | 58 | 27 | QA | −1 | |
| Top genes associated with young group (<50 years old) versus old (285). | 5TGM7 | 0.002186 | 4 | 4 | 85 | 43 | QA | −1 |
| MRE11A | 0.006959 | 8 | 8 | 177 | 62 | QA | −1 | |
| NBN | 0.011436 | 11 | 11 | 171 | 47 | QA | −1 | |
| VHL | 0.015733 | 2 | 2 | 2 | 2 | ER | 0.015733 | |
| IDH1 | 0.019368 | 7 | 7 | 16 | 12 | ER | 1.32 × 10−10 | |
| Top genes associated with left colorectal cancer versus right (204). | PIK3CB | 0.0007 | 9 | 9 | 9 | 8 | ER | 8.76 × 10−5 |
| PIK3CA | 0.001 | 12 | 12 | 86 | 39 | QA | −1 | |
| RNF213 | 0.001 | 61 | 56 | 709 | 78 | MA | −1 | |
| AURKB | 0.003 | 5 | 5 | 101 | 39 | QA | −1 | |
| ERBB4 | 0.005 | 11 | 11 | 41 | 27 | QA | −1 | |
| Top genes associated with late stage versus early stage (180). | EXT1 | 0.003027 | 7 | 7 | 73 | 51 | MA | −1 |
| RNASEL | 0.013275 | 4 | 4 | 34 | 29 | ER.A | −1 | |
| CDH5 | 0.018248 | 8 | 8 | 135 | 62 | MA | −1 | |
| BUB1B | 0.021125 | 18 | 18 | 178 | 48 | MA | −1 | |
| MUTYH | 0.027108 | 11 | 11 | 108 | 51 | MA | −1 |
| Symbol | Entrez Gene Name | Location | Family | Entrez Gene ID for Human |
|---|---|---|---|---|
| APC | APC regulator of WNT signaling pathway | Nucleus | Enzyme | 324 |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase | Cytoplasm | Kinase | 673 |
| CTNNB1 | Catenin beta 1 | Nucleus | Transcription regulator | 1499 |
| EGFR | epidermal growth factor receptor | Plasma membrane | Kinase | 1956 |
| ERBB2 | Erb-b2 receptor tyrosine kinase 2 | Plasma membrane | Kinase | 2064 |
| FLT1 | Fms-related receptor tyrosine kinase 1 | Plasma membrane | Kinase | 2321 |
| IGF1R | Insulin-like growth factor 1 receptor | Plasma membrane | Transmembrane receptor | 3480 |
| KDR | Kinase insert domain receptor | Plasma membrane | Kinase | 3791 |
| KIT | KIT proto-oncogene, receptor tyrosine kinase | Plasma membrane | Transmembrane receptor | 3815 |
| KRAS | KRAS proto-oncogene, GTPase | Cytoplasm | Enzyme | 3845 |
| MLH1 | MutL homolog 1 | Nucleus | Enzyme | 4292 |
| PDGFRA | Platelet-derived growth factor receptor alpha | Plasma membrane | Kinase | 5156 |
| PIK3CA | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha | Cytoplasm | Kinase | 5290 |
| PTEN | Phosphatase and tensin homolog | Cytoplasm | Phosphatase | 5728 |
| SMAD4 | SMAD family member 4 | Nucleus | Transcription regulator | 4089 |
| TP53 | Tumor protein p53 | Nucleus | Transcription regulator | 7157 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almuzzaini, B.; Alghamdi, J.; Alomani, A.; AlGhamdi, S.; Alsharm, A.A.; Alshieban, S.; Sayed, A.; Alhejaily, A.G.; Aljaser, F.S.; Abudawood, M.; et al. Identification of Novel Mutations in Colorectal Cancer Patients Using AmpliSeq Comprehensive Cancer Panel. J. Pers. Med. 2021, 11, 535. https://doi.org/10.3390/jpm11060535
Almuzzaini B, Alghamdi J, Alomani A, AlGhamdi S, Alsharm AA, Alshieban S, Sayed A, Alhejaily AG, Aljaser FS, Abudawood M, et al. Identification of Novel Mutations in Colorectal Cancer Patients Using AmpliSeq Comprehensive Cancer Panel. Journal of Personalized Medicine. 2021; 11(6):535. https://doi.org/10.3390/jpm11060535
Chicago/Turabian StyleAlmuzzaini, Bader, Jahad Alghamdi, Alhanouf Alomani, Saleh AlGhamdi, Abdullah A. Alsharm, Saeed Alshieban, Ahood Sayed, Abdulmohsen G. Alhejaily, Feda S. Aljaser, Manal Abudawood, and et al. 2021. "Identification of Novel Mutations in Colorectal Cancer Patients Using AmpliSeq Comprehensive Cancer Panel" Journal of Personalized Medicine 11, no. 6: 535. https://doi.org/10.3390/jpm11060535
APA StyleAlmuzzaini, B., Alghamdi, J., Alomani, A., AlGhamdi, S., Alsharm, A. A., Alshieban, S., Sayed, A., Alhejaily, A. G., Aljaser, F. S., Abudawood, M., Almajed, F., Samman, A., Balwi, M. A. A., & Aziz, M. A. (2021). Identification of Novel Mutations in Colorectal Cancer Patients Using AmpliSeq Comprehensive Cancer Panel. Journal of Personalized Medicine, 11(6), 535. https://doi.org/10.3390/jpm11060535