
A Type 2 Ryanodine Receptor Variant in the Helical Domain 2 Associated with an Impairment of the Adrenergic Response

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Genetic Analysis
2.3. RYR2 Cloning and Mutagenesis
2.4. Cell Culture and Transfection
2.5. Protein Extraction and Western Blotting
2.6. Calcium Imaging
2.7. Single Channel Analysis
2.8. Statistical Analysis
3. Results
3.1. Clinical Phenotype and Genetic Analysis
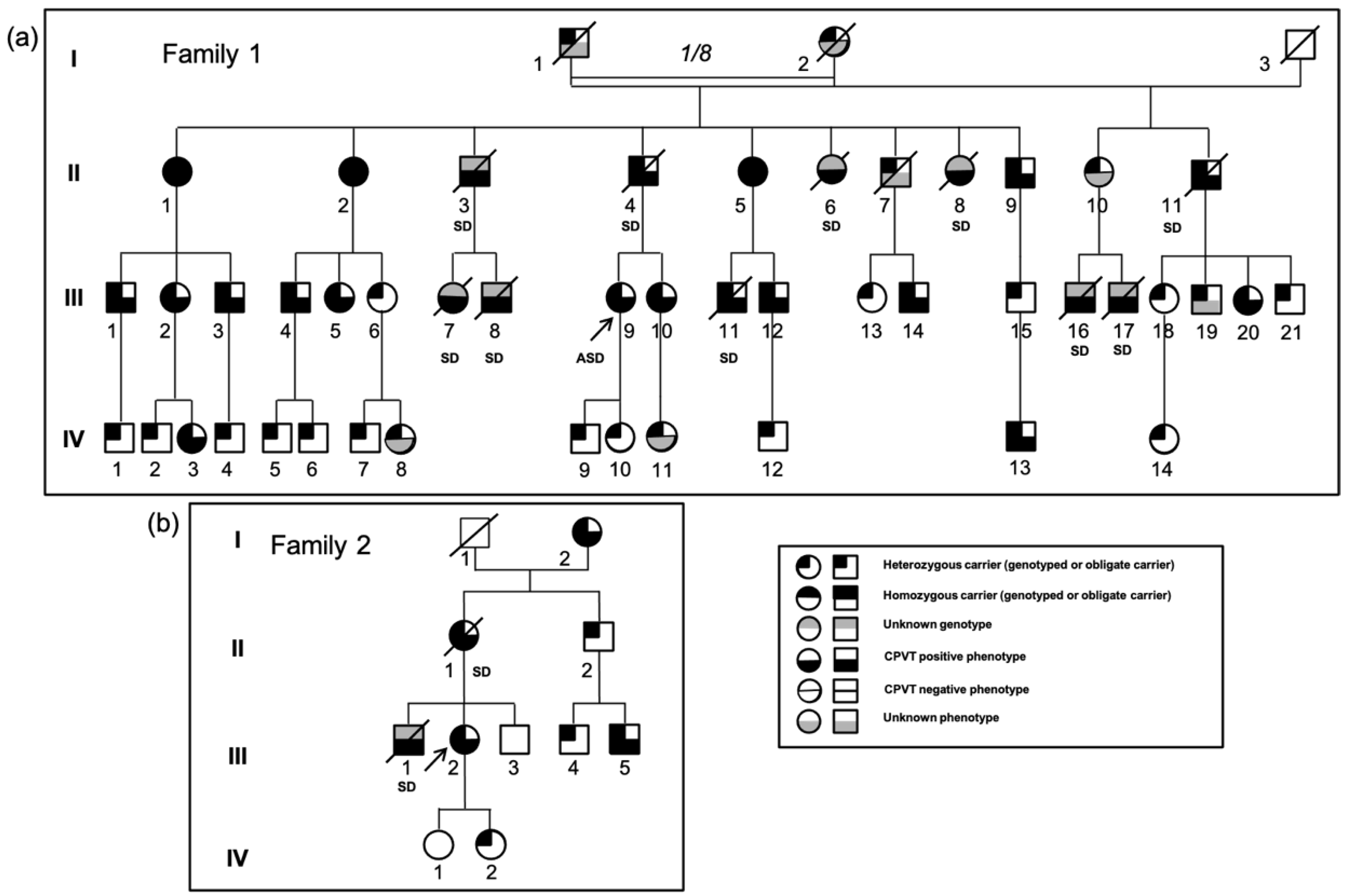
3.1.1. Family 1
3.1.2. Family 2
3.1.3. Family 3
3.2. Position and Conservation of the RyR2 D3291V Variant
3.3. Blunted Adrenergic Response of D3291V-RyR2 Channels in HEK293 Cells
3.4. Decreased Phosphorylation Level of D3291V Channels at S2808
3.5. Dominant Effect of D3291V Variant in HL-1 Cells
3.6. Loss of Luminal Ca2+ Sensitivity in D3291V-RyR2 Channels
4. Discussion
4.1. Clinical Characterization of the Families
4.2. Identification of a Novel RYR2 Variant
4.3. Functional Study of the D3291V-RyR2 Channels
5. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Leenhardt, A.; Lucet, V.; Denjoy, I.; Grau, F.; Ngoc, D.D.; Coumel, P. Catecholaminergic Polymorphic Ventricular Tachycardia in Children. Circulation 1995, 91, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Napolitano, C.; Priori, S.G. Catecholaminergic Polymorphic Ventricular Tachycardia: A Paradigm to Understand Mechanisms of Arrhythmias Associated to Impaired Ca2+ Regulation. Heart Rhythm 2009, 6, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac DeathThe Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.; Denjoy, I.; Extramiana, F.; Maltret, A.; Buisson, N.R.; Lupoglazoff, J.-M.; Klug, D.; Hayashi, M.; Takatsuki, S.; Villain, E.; et al. Incidence and Risk Factors of Arrhythmic Events in Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2009, 119, 2426–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and Molecular Characterization of Patients With Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef] [Green Version]
- van der Werf, C.; Nederend, I.; Hofman, N.; van Geloven, N.; Ebink, C.; Frohn-Mulder, I.M.E.; Alings, A.M.W.; Bosker, H.A.; Bracke, F.A.; van den Heuvel, F.; et al. Familial Evaluation in Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Arrhythmia Electrophysiol. 2012, 5, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Kannankeril, P.J.; Moore, J.P.; Cerrone, M.; Priori, S.G.; Kertesz, N.J.; Ro, P.S.; Batra, A.S.; Kaufman, E.S.; Fairbrother, D.L.; Saarel, E.V.; et al. Efficacy of Flecainide in the Treatment of Catecholaminergic Polymorphic Ventricular Tachycardia: A Randomized Clinical Trial. JAMA Cardiol. 2017, 2, 759–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, H.; Chopra, N.; Laver, D.; Hwang, H.S.; Davies, S.S.; Roach, D.E.; Duff, H.J.; Roden, D.M.; Wilde, A.A.M.; Knollmann, B.C. Flecainide Prevents Catecholaminergic Polymorphic Ventricular Tachycardia in Mice and Humans. Nat. Med. 2009, 15, 380–383. [Google Scholar] [CrossRef]
- Laitinen, P.J.; Brown, K.M.; Piippo, K.; Swan, H.; Devaney, J.M.; Brahmbhatt, B.; Donarum, E.A.; Marino, M.; Tiso, N.; Viitasalo, M.; et al. Mutations of the Cardiac Ryanodine Receptor (RyR2) Gene in Familial Polymorphic Ventricular Tachycardia. Circulation 2001, 103, 485–490. [Google Scholar] [CrossRef]
- Medeiros-Domingo, A.; Bhuiyan, Z.A.; Tester, D.J.; Hofman, N.; Bikker, H.; van Tintelen, J.P.; Mannens, M.M.A.M.; Wilde, A.A.M.; Ackerman, M.J. Comprehensive Open Reading Frame Mutational Analysis of the RYR2-Encoded Ryanodine Receptor/Calcium Channel in Patients Diagnosed Previously with Either Catecholaminergic Polymorphic Ventricular Tachycardia or Genotype Negative, Exercise-Induced Long QT Syndrome. J. Am. Coll. Cardiol. 2009, 54, 2065–2074. [Google Scholar] [CrossRef] [Green Version]
- Postma, A.; Denjoy, I.; Kamblock, J.; Alders, M.; Lupoglazoff, J.; Vaksmann, G.; Dubosq-Bidot, L.; Sebillon, P.; Mannens, M.; Guicheney, P.; et al. Catecholaminergic Polymorphic Ventricular Tachycardia: RYR2 Mutations, Bradycardia, and Follow up of the Patients. J. Med. Genet. 2005, 42, 863–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the Cardiac Ryanodine Receptor Gene (HRyR2) Underlie Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leenhardt, A.; Denjoy, I.; Guicheney, P. Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Arrhythmia Electrophysiol. 2012, 5, 1044–1052. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Hurtado, N.; Boczek, N.J.; Kryshtal, D.O.; Johnson, C.N.; Sun, J.; Nitu, F.R.; Cornea, R.L.; Chazin, W.J.; Calvert, M.L.; Tester, D.J.; et al. Novel CPVT-Associated Calmodulin Mutation in CALM3 (CALM3-A103V) Activates Arrhythmogenic Ca Waves and Sparks. Circ. Arrhythmia Electrophysiol. 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Hwang, H.S.; Nitu, F.R.; Yang, Y.; Walweel, K.; Pereira, L.; Johnson, C.N.; Faggioni, M.; Chazin, W.J.; Laver, D.; George, A.L.; et al. Divergent Regulation of Ryanodine Receptor 2 Calcium Release Channels by Arrhythmogenic Human Calmodulin Missense Mutants. Circ. Res. 2014, 114, 1114–1124. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Jáimez, J.; Palomino Doza, J.; Ortega, Á.; Macías-Ruiz, R.; Perin, F.; Rodríguez-Vázquez del Rey, M.M.; Ortiz-Genga, M.; Monserrat, L.; Barriales-Villa, R.; Blanca, E.; et al. Calmodulin 2 Mutation N98S Is Associated with Unexplained Cardiac Arrest in Infants Due to Low Clinical Penetrance Electrical Disorders. PLoS ONE 2016, 11, e0153851. [Google Scholar] [CrossRef] [PubMed]
- Makita, N.; Yagihara, N.; Crotti, L.; Johnson, C.N.; Beckmann, B.-M.; Roh, M.S.; Shigemizu, D.; Lichtner, P.; Ishikawa, T.; Aiba, T.; et al. Novel Calmodulin Mutations Associated with Congenital Arrhythmia Susceptibility. Circ. Cardiovasc. Genet. 2014, 7, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Nyegaard, M.; Overgaard, M.T.; Søndergaard, M.T.; Vranas, M.; Behr, E.R.; Hildebrandt, L.L.; Lund, J.; Hedley, P.L.; Camm, A.J.; Wettrell, G.; et al. Mutations in Calmodulin Cause Ventricular Tachycardia and Sudden Cardiac Death. Am. J. Hum. Genet. 2012, 91, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A Missense Mutation in a Highly Conserved Region of CASQ2 Is Associated with Autosomal Recessive Catecholamine-Induced Polymorphic Ventricular Tachycardia in Bedouin Families from Israel. Am. J. Hum. Genet. 2001, 69, 1378–1384. [Google Scholar] [CrossRef] [Green Version]
- Postma, A.V.; Denjoy, I.; Hoorntje, T.M.; Lupoglazoff, J.-M.; Costa, A.D.; Sebillon, P.; Mannens, M.M.A.M.; Wilde, A.A.M.; Guicheney, P. Absence of Calsequestrin 2 Causes Severe Forms of Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2002, 91, e21–e26. [Google Scholar] [CrossRef]
- Roux-Buisson, N.; Cacheux, M.; Fourest-Lieuvin, A.; Fauconnier, J.; Brocard, J.; Denjoy, I.; Durand, P.; Guicheney, P.; Kyndt, F.; Leenhardt, A.; et al. Absence of Triadin, a Protein of the Calcium Release Complex, Is Responsible for Cardiac Arrhythmia with Sudden Death in Human. Hum. Mol. Genet. 2012, 21, 2759–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Xiao, B.; Zhang, L.; Chen, S.R. Wayne Enhanced Basal Activity of a Cardiac Ca2+ Release Channel (Ryanodine Receptor) Mutant Associated With Ventricular Tachycardia and Sudden Death. Circ. Res. 2002, 91, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Xiao, B.; Yang, D.; Wang, R.; Choi, P.; Zhang, L.; Cheng, H.; Chen, S.R.W. RyR2 Mutations Linked to Ventricular Tachycardia and Sudden Death Reduce the Threshold for Store-Overload-Induced Ca2+ Release (SOICR). Proc. Natl. Acad. Sci. USA 2004, 101, 13062–13067. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Wang, R.; Xiao, B.; Kong, H.; Hunt, D.J.; Choi, P.; Zhang, L.; Chen, S.R.W. Enhanced Store Overload-Induced Ca2+ Release and Channel Sensitivity to Luminal Ca2+ Activation Are Common Defects of RyR2 Mutations Linked to Ventricular Tachycardia and Sudden Death. Circ. Res. 2005, 97, 1173–1181. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Knollmann, B.C. Mechanism Underlying Catecholaminergic Polymorphic Ventricular Tachycardia and Approaches to Therapy. J. Electrocardiol. 2011, 44, 650–655. [Google Scholar] [CrossRef]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of Mutations in the Cardiac Ryanodine Receptor Gene in Families Affected with Arrhythmogenic Right Ventricular Cardiomyopathy Type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roston, T.M.; Guo, W.; Krahn, A.D.; Wang, R.; Van Petegem, F.; Sanatani, S.; Chen, S.R.W.; Lehman, A. A Novel RYR2 Loss-of-Function Mutation (I4855M) Is Associated with Left Ventricular Non-Compaction and Atypical Catecholaminergic Polymorphic Ventricular Tachycardia. J. Electrocardiol. 2017, 50, 227–233. [Google Scholar] [CrossRef] [PubMed]
- George, C.H.; Jundi, H.; Thomas, N.L.; Fry, D.L.; Lai, F.A. Ryanodine Receptors and Ventricular Arrhythmias: Emerging Trends in Mutations, Mechanisms and Therapies. J. Mol. Cell. Cardiol. 2007, 42, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Venetucci, L.; Denegri, M.; Napolitano, C.; Priori, S.G. Inherited Calcium Channelopathies in the Pathophysiology of Arrhythmias. Nat. Rev. Cardiol. 2012, 9, 561. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Itoh, H.; Ohno, S.; Murayama, T.; Kurebayashi, N.; Aoki, H.; Blancard, M.; Nakagawa, Y.; Yamamoto, S.; Matsui, Y.; et al. A Type 2 Ryanodine Receptor Variant Associated with Reduced Ca2+ Release and Short-Coupled Torsades de Pointes Ventricular Arrhythmia. Heart Rhythm 2017, 14, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Yao, J.; Ni, M.; Wei, J.; Zhong, X.; Guo, W.; Zhang, L.; Wang, R.; Belke, D.; Chen, Y.-X.; et al. Cardiac Ryanodine Receptor Calcium Release Deficiency Syndrome. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Chen, W.; Wang, R.; Zhang, L.; Chen, S.R.W. Loss of Luminal Ca2+ Activation in the Cardiac Ryanodine Receptor Is Associated with Ventricular Fibrillation and Sudden Death. Proc. Natl. Acad. Sci. USA 2007, 104, 18309–18314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Guo, W.; Wei, J.; Tang, Y.; Liu, Y.; Zhang, J.Z.; Tan, V.H.; Zhang, L.; Wang, R.; Jones, P.P.; et al. Identification of Loss-of-Function RyR2 Mutations Associated with Idiopathic Ventricular Fibrillation and Sudden Death. Biosci. Rep. 2021, 41. [Google Scholar] [CrossRef]
- Paech, C.; Gebauer, R.A.; Karstedt, J.; Marschall, C.; Bollmann, A.; Husser, D. Ryanodine Receptor Mutations Presenting as Idiopathic Ventricular Fibrillation: A Report on Two Novel Familial Compound Mutations, c.6224T>C and c.13781A>G, With the Clinical Presentation of Idiopathic Ventricular Fibrillation. Pediatr. Cardiol. 2014, 35, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Tunwell, R.E.; Wickenden, C.; Bertrand, B.M.; Shevchenko, V.I.; Walsh, M.B.; Allen, P.D.; Lai, F.A. The Human Cardiac Muscle Ryanodine Receptor-Calcium Release Channel: Identification, Primary Structure and Topological Analysis. Biochem. J. 1996, 318, 477–487. [Google Scholar] [CrossRef] [Green Version]
- George, C.H.; Sorathia, R.; Bertrand, B.M.A.; Lai, F.A. In Situ Modulation of the Human Cardiac Ryanodine Receptor (HRyR2) by FKBP12.6. Biochem. J. 2003, 370, 579–589. [Google Scholar] [CrossRef]
- Touat-Hamici, Z.; Blancard, M.; Ma, R.; Lin, L.; Iddir, Y.; Denjoy, I.; Leenhardt, A.; Yuchi, Z.; Guicheney, P. A SPRY1 Domain Cardiac Ryanodine Receptor Variant Associated with Short-Coupled Torsade de Pointes. Sci. Rep. 2021, 11, 5243. [Google Scholar] [CrossRef]
- Peng, W.; Shen, H.; Wu, J.; Guo, W.; Pan, X.; Wang, R.; Chen, S.R.W.; Yan, N. Structural Basis for the Gating Mechanism of the Type 2 Ryanodine Receptor RyR2. Science 2016, 354, aah5324. [Google Scholar] [CrossRef] [PubMed]
- Yuchi, Z.; Van Petegem, F. Ryanodine Receptors under the Magnifying Lens: Insights and Limitations of Cryo-Electron Microscopy and X-Ray Crystallography Studies. Cell Calcium 2016, 59, 209–227. [Google Scholar] [CrossRef]
- Yan, Z.; Bai, X.; Yan, C.; Wu, J.; Li, Z.; Xie, T.; Peng, W.; Yin, C.; Li, X.; Scheres, S.H.W.; et al. Structure of the Rabbit Ryanodine Receptor RyR1 at Near-Atomic Resolution. Nature 2015, 517, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Marjamaa, A.; Laitinen-Forsblom, P.; Lahtinen, A.M.; Viitasalo, M.; Toivonen, L.; Kontula, K.; Swan, H. Search for Cardiac Calcium Cycling Gene Mutations in Familial Ventricular Arrhythmias Resembling Catecholaminergic Polymorphic Ventricular Tachycardia. BMC Med. Genet. 2009, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Claycomb, W.C.; Lanson, N.A.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J. HL-1 Cells: A Cardiac Muscle Cell Line That Contracts and Retains Phenotypic Characteristics of the Adult Cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauce, B.; Rampazzo, A.; Basso, C.; Bagattin, A.; Daliento, L.; Tiso, N.; Turrini, P.; Thiene, G.; Danieli, G.A.; Nava, A. Screening for Ryanodine Receptor Type 2 Mutations in Families with Effort-Induced Polymorphic Ventricular Arrhythmias and Sudden Death: Early Diagnosis of Asymptomatic Carriers. J. Am. Coll. Cardiol. 2002, 40, 341–349. [Google Scholar] [CrossRef]
- Swan, H.; Piippo, K.; Viitasalo, M.; Heikkilä, P.; Paavonen, T.; Kainulainen, K.; Kere, J.; Keto, P.; Kontula, K.; Toivonen, L. Arrhythmic Disorder Mapped to Chromosome 1q42–Q43 Causes Malignant Polymorphic Ventricular Tachycardia in Structurally Normal Hearts. J. Am. Coll. Cardiol. 1999, 34, 2035–2042. [Google Scholar] [CrossRef] [Green Version]
- Sy, R.W.; Gollob, M.H.; Klein, G.J.; Yee, R.; Skanes, A.C.; Gula, L.J.; Leong-Sit, P.; Gow, R.M.; Green, M.S.; Birnie, D.H.; et al. Arrhythmia Characterization and Long-Term Outcomes in Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Rhythm 2011, 8, 864–871. [Google Scholar] [CrossRef]
- Milting, H.; Lukas, N.; Klauke, B.; Körfer, R.; Perrot, A.; Osterziel, K.-J.; Vogt, J.; Peters, S.; Thieleczek, R.; Varsányi, M. Composite Polymorphisms in the Ryanodine Receptor 2 Gene Associated with Arrhythmogenic Right Ventricular Cardiomyopathy. Cardiovasc. Res. 2006, 71, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Atik, S.U.; Alp, F.E.; Dedeoğlu, R.; Koka, A.; Öztunç, F.; Eroğlu, A.G. A Rare Cause of Sudden Cardiac Arrest: Catecholaminergic Polymorphic Ventricular Tachycardia. Turk. Pediatri. Ars. 2018, 53, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Broendberg, A.K.; Nielsen, J.C.; Bjerre, J.; Pedersen, L.N.; Kristensen, J.; Henriksen, F.L.; Bundgaard, H.; Jensen, H.K. Nationwide Experience of Catecholaminergic Polymorphic Ventricular Tachycardia Caused by RyR2 Mutations. Heart 2017, 103, 901–909. [Google Scholar] [CrossRef]
- Amburgey, K.; Bailey, A.; Hwang, J.H.; Tarnopolsky, M.A.; Bonnemann, C.G.; Medne, L.; Mathews, K.D.; Collins, J.; Daube, J.R.; Wellman, G.P.; et al. Genotype-Phenotype Correlations in Recessive RYR1-Related Myopathies. Orphanet J. Rare Dis. 2013, 8, 117. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Jones, P.P.; Davis, D.R.; Gow, R.; Green, M.S.; Birnie, D.H.; Chen, S.R.W.; Gollob, M.H. Characterization of a Novel Mutation in the Cardiac Ryanodine Receptor That Results in Catecholaminergic Polymorphic Ventricular Tachycardia. Channels 2010, 4, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, N.L.; George, C.H.; Lai, F.A. Functional Heterogeneity of Ryanodine Receptor Mutations Associated with Sudden Cardiac Death. Cardiovasc. Res. 2004, 64, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangüemert, F.; Bosch Calero, C.; Pérez, C.; Campuzano, O.; Beltran-Alvarez, P.; Scornik, F.S.; Iglesias, A.; Berne, P.; Allegue, C.; Ruiz Hernandez, P.M.; et al. Clinical and Molecular Characterization of a Cardiac Ryanodine Receptor Founder Mutation Causing Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Rhythm 2015, 12, 1636–1643. [Google Scholar] [CrossRef] [Green Version]
- Meissner, G. The Structural Basis of Ryanodine Receptor Ion Channel Function. J. Gen. Physiol. 2017, 149, 1065–1089. [Google Scholar] [CrossRef] [PubMed]
- Haji-Ghassemi, O.; Yuchi, Z.; Van Petegem, F. The Cardiac Ryanodine Receptor Phosphorylation Hotspot Embraces PKA in a Phosphorylation-Dependent Manner. Mol. Cell 2019, 75, 39–52.e4. [Google Scholar] [CrossRef]
- Ogawa, H.; Kurebayashi, N.; Yamazawa, T.; Murayama, T. Regulatory Mechanisms of Ryanodine Receptor/Ca2+ Release Channel Revealed by Recent Advancements in Structural Studies. J. Muscle Res. Cell Motil. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efremov, R.G.; Leitner, A.; Aebersold, R.; Raunser, S. Architecture and Conformational Switch Mechanism of the Ryanodine Receptor. Nature 2015, 517, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Zalk, R.; Clarke, O.B.; des Georges, A.; Grassucci, R.A.; Reiken, S.; Mancia, F.; Hendrickson, W.A.; Frank, J.; Marks, A.R. Structure of a Mammalian Ryanodine Receptor. Nature 2015, 517, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laver, D.R. Ca2+ Stores Regulate Ryanodine Receptor Ca2+ Release Channels via Luminal and Cytosolic Ca2+ Sites. Biophys. J. 2007, 92, 3541–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fill, M.; Copello, J.A. Ryanodine Receptor Calcium Release Channels. Physiol. Rev. 2002, 82, 893–922. [Google Scholar] [CrossRef] [Green Version]
- Györke, I.; Györke, S. Regulation of the Cardiac Ryanodine Receptor Channel by Luminal Ca2+ Involves Luminal Ca2+ Sensing Sites. Biophys. J. 1998, 75, 2801–2810. [Google Scholar] [CrossRef] [Green Version]
- Porta, M.; Zima, A.V.; Nani, A.; Diaz-Sylvester, P.L.; Copello, J.A.; Ramos-Franco, J.; Blatter, L.A.; Fill, M. Single Ryanodine Receptor Channel Basis of Caffeine’s Action on Ca2+ Sparks. Biophys. J. 2011, 100, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Györke, S.; Terentyev, D. Modulation of Ryanodine Receptor by Luminal Calcium and Accessory Proteins in Health and Cardiac Disease. Cardiovasc. Res. 2008, 77, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-T.; Valdivia, C.R.; Gurrola, G.B.; Powers, P.P.; Willis, B.C.; Moss, R.L.; Jalife, J.; Valdivia, H.H. Arrhythmogenesis in a Catecholaminergic Polymorphic Ventricular Tachycardia Mutation That Depresses Ryanodine Receptor Function. Proc. Natl. Acad. Sci. USA 2015, 112, E1669–E1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleiman, Y.; Souidi, M.; Kumar, R.; Yang, E.; Jaffré, F.; Zhou, T.; Bernardin, A.; Reiken, S.; Cazorla, O.; Kajava, A.V.; et al. Modeling Polymorphic Ventricular Tachycardia at Rest Using Patient-Specific Induced Pluripotent Stem Cell-Derived Cardiomyocytes. EBioMedicine 2020, 60. [Google Scholar] [CrossRef]
- Acimovic, I.; Refaat, M.M.; Moreau, A.; Salykin, A.; Reiken, S.; Sleiman, Y.; Souidi, M.; Přibyl, J.; Kajava, A.V.; Richard, S.; et al. Post-Translational Modifications and Diastolic Calcium Leak Associated to the Novel RyR2-D3638A Mutation Lead to CPVT in Patient-Specific HiPSC-Derived Cardiomyocytes. J. Clin. Med. 2018, 7, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blancard, M.; Touat-Hamici, Z.; Aguilar-Sanchez, Y.; Yin, L.; Vaksmann, G.; Roux-Buisson, N.; Fressart, V.; Denjoy, I.; Klug, D.; Neyroud, N.; et al. A Type 2 Ryanodine Receptor Variant in the Helical Domain 2 Associated with an Impairment of the Adrenergic Response. J. Pers. Med. 2021, 11, 579. https://doi.org/10.3390/jpm11060579
Blancard M, Touat-Hamici Z, Aguilar-Sanchez Y, Yin L, Vaksmann G, Roux-Buisson N, Fressart V, Denjoy I, Klug D, Neyroud N, et al. A Type 2 Ryanodine Receptor Variant in the Helical Domain 2 Associated with an Impairment of the Adrenergic Response. Journal of Personalized Medicine. 2021; 11(6):579. https://doi.org/10.3390/jpm11060579
Chicago/Turabian StyleBlancard, Malorie, Zahia Touat-Hamici, Yuriana Aguilar-Sanchez, Liheng Yin, Guy Vaksmann, Nathalie Roux-Buisson, Véronique Fressart, Isabelle Denjoy, Didier Klug, Nathalie Neyroud, and et al. 2021. "A Type 2 Ryanodine Receptor Variant in the Helical Domain 2 Associated with an Impairment of the Adrenergic Response" Journal of Personalized Medicine 11, no. 6: 579. https://doi.org/10.3390/jpm11060579