
The Skeletal Muscle Emerges as a New Disease Target in Amyotrophic Lateral Sclerosis

 , , , ,
, , , , 
Abstract
:
1. Introduction
2. Motor Neuron Degeneration as Non-Cell Autonomous Process
2.1. Microglia
2.2. Astrocytes
2.3. Skeletal Muscle Cells
3. The Skeletal Muscle in ALS Context
3.1. Specific Mouse Models Used in ALS Research
3.2. Muscle Atrophy in Disease
3.3. Perturbations of Energy Metabolism in ALS Muscle
3.3.1. Muscle Exercising and The Risk of ALS
3.3.2. Alterations of Muscle Metabolism as Prodromal Features of ALS
4. Pathological Spreading into Neurons: Mechanisms
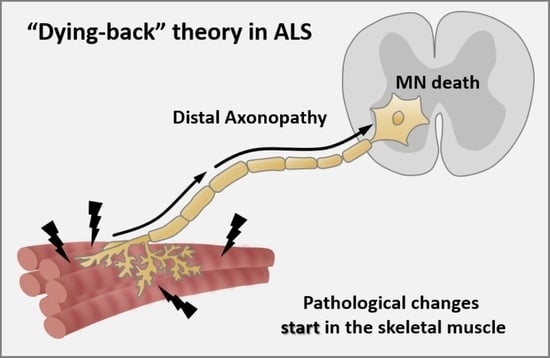
Axonopathy—The Axonal Degeneration
5. The Neuromuscular Junction in ALS
5.1. Pre-Synaptic Affectation
5.2. Post-Synaptic Affectation
6. Final Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morrison, B.M. Neuromuscular Diseases. Semin. Neurol. 2016, 36, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Ehmsen, J.T.; Höke, A. Cellular and molecular features of neurogenic skeletal muscle atrophy. Exp. Neurol. 2020, 331, 113379. [Google Scholar] [CrossRef] [PubMed]
- Pandya, V.A.; Patani, R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: A cellular perspective. Brain 2020, 143, 1057–1072. [Google Scholar] [CrossRef] [PubMed]
- Hinchcliffe, M.; Smith, A. Riluzole: real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener. Neurol. Neuromuscul. Dis. 2017, Volume 7, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Shukla, S. Role of edaravone as a treatment option for patients with amyotrophic lateral sclerosis. Pharmaceuticals 2021, 14, 29. [Google Scholar] [CrossRef]
- Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. DMM Dis. Model. Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zufiría, M.; Gil-Bea, F.J.; Fernández-Torrón, R.; Poza, J.J.; Muñoz-Blanco, J.L.; Rojas-García, R.; Riancho, J.; de Munain, A.L. ALS: A bucket of genes, environment, metabolism and unknown ingredients. Prog. Neurobiol. 2016, 142, 104–129. [Google Scholar] [CrossRef]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G. Le Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Lafarga, V.; Sirozh, O.; Díaz-López, I.; Galarreta, A.; Hisaoka, M.; Zarzuela, E.; Boskovic, J.; Jovanovic, B.; Fernandez-Leiro, R.; Muñoz, J.; et al. Widespread displacement of DNA- and RNA-binding factors underlies toxicity of arginine-rich cell-penetrating peptides. EMBO J. 2021, 1–16. [Google Scholar] [CrossRef]
- Tsitkanou, S.; Gatta, P.A.D.; Russell, A.P. Skeletal muscle satellite cells, mitochondria, and MicroRNAs: Their involvement in the pathogenesis of ALS. Front. Physiol. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, D.; Pasetto, L.; Bonetto, V.; Basso, M. Role of Extracellular Vesicles in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2018, 12, 1–9. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y.; Ryu, H. Astrocytes and microglia as non-cell autonomous players in the pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Chand, K.K.; Lee, K.M.; Lee, J.D.; Qiu, H.; Willis, E.F.; Lavidis, N.A.; Hilliard, M.A.; Noakes, P.G. Defects in synaptic transmission at the neuromuscular junction precede motor deficits in a TDP-43Q331K transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 2018, 32, 2676–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. ALS as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 2014, 8, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallon, C.; Russell, K.A.; Sakhalkar, S.; Andrapallayal, N.; Farah, M.H. Length-dependent axo-terminal degeneration at the neuromuscular synapses of type II muscle in SOD1 mice. Neuroscience 2016, 312, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsumi, R.; Sankoda, Y.; Anderson, J.E.; Sato, Y.; Mizunoya, W.; Shimizu, N.; Suzuki, T.; Yamada, M.; Rhoads, R.P.; Ikeuchi, Y.; et al. Possible implication of satellite cells in regenerative motoneuritogenesis: HGF upregulates neural chemorepellent Sema3A during myogenic differentiation. Am. J. Physiol. Cell Physiol. 2009, 297, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Boncompagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal Muscle Is a Primary Target of SOD1G93A-Mediated Toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Wong, M. Skeletal Muscle-Restricted Expression of Human SOD1 in Transgenic Mice Causes a Fatal ALS-Like Syndrome. Front. Neurol. 2020, 11, 1–26. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Drey, M.; Krieger, B.; Sieber, C.C.; Bauer, J.M.; Hettwer, S.; Bertsch, T. Motoneuron loss is associated with sarcopenia. J. Am. Med. Dir. Assoc. 2014, 15, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Arbour, D.; Vande Velde, C.; Robitaille, R. New perspectives on amyotrophic lateral sclerosis: The role of glial cells at the neuromuscular junction. J. Physiol. 2017, 595, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Vastano, R.; Perez, M.A. Changes in motoneuron excitability during voluntary muscle activity in humans with spinal cord injury. J. Neurophysiol. 2020, 123, 454–461. [Google Scholar] [CrossRef]
- Manzano, R.; Toivonen, J.M.; Moreno-Martínez, L.; de la Torre, M.; Moreno-García, L.; López-Royo, T.; Molina, N.; Zaragoza, P.; Calvo, A.C.; Osta, R. What skeletal muscle has to say in amyotrophic lateral sclerosis: Implications for therapy. Br. J. Pharmacol. 2021, 178, 1279–1297. [Google Scholar] [CrossRef] [PubMed]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Puentes, F.; Malaspina, A.; Van Noort, J.M.; Amor, S. Non-neuronal cells in ALS: Role of glial, immune cells and blood-CNS barriers. In Proceedings of the Brain Pathology; Blackwell Publishing Ltd.: Oxford, UK, 2016; Volume 26, pp. 248–257. [Google Scholar]
- Appel, S.H.; Smith, R.G.; Engelhardt, J.I.; Stefani, E. Evidence for autoimmunity in amyotrophic lateral sclerosis. J. Neurol. Sci. 1993, 118, 169–174. [Google Scholar] [CrossRef]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2013, 5, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [Green Version]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and microglia: Novel partners in neurodegeneration and aging. Int. J. Mol. Sci. 2017, 18, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist. Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Zürcher, N.R.; Loggia, M.L.; Lawson, R.; Chonde, D.B.; Izquierdo-Garcia, D.; Yasek, J.E.; Akeju, O.; Catana, C.; Rosen, B.R.; Cudkowicz, M.E.; et al. Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: Assessed with [11C]-PBR28. NeuroImage Clin. 2015, 7, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, J.S.; Engelhardt, J.I.; Siklós, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of Dendritic Cells, MCP-1, and Activated Microglia/Macrophages in Amyotrophic Lateral Sclerosis Spinal Cord Tissue. Ann. Neurol. 2004, 55, 221–235. [Google Scholar] [CrossRef]
- Brettschneider, J.; Toledo, J.B.; van Deerlin, V.M.; Elman, L.; McCluskey, L.; Lee, V.M.Y.; Trojanowski, J.Q. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e39216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Rodent models of amyotrophic lateral sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5–67. [Google Scholar] [CrossRef] [Green Version]
- Gravel, M.; Béland, L.C.; Soucy, G.; Abdelhamid, E.; Rahimian, R.; Gravel, C.; Kriz, J. Il-10 controls early microglial phenotypes and disease onset in ALS caused by misfolded superoxide dismutase 1. J. Neurosci. 2016, 36, 1031–1048. [Google Scholar] [CrossRef]
- Pramatarova, A.; Laganière, J.; Roussel, J.; Brisebois, K.; Rouleau, G.A. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 2001, 21, 3369–3374. [Google Scholar] [CrossRef] [Green Version]
- Lino, M.M.; Schneider, C.; Caroni, P. Accumulation of SOD1 Mutants in Postnatal Motoneurons Does Not Cause Motoneuron Pathology or Motoneuron Disease. J. Neurosci. 2002, 22, 4825–4832. [Google Scholar] [CrossRef] [Green Version]
- Vahsen, B.F.; Gray, E.; Thompson, A.G.; Ansorge, O.; Anthony, D.C.; Cowley, S.A.; Talbot, K.; Turner, M.R. Non-neuronal cells in amyotrophic lateral sclerosis — from pathogenesis to biomarkers. Nat. Rev. Neurol. 2021, 17, 333–348. [Google Scholar] [CrossRef]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Sharma, K.; Deng, H.X.; Siddique, T.; Grisotti, G.; Liu, E.; Roos, R.P. Restricted expression of mutant SOD1 in spinal motor neurons and interneurons induces motor neuron pathology. Neurobiol. Dis. 2008, 29, 400–408. [Google Scholar] [CrossRef]
- Liao, B.; Zhao, W.; Beers, D.R.; Henkel, J.S.; Appel, S.H. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 2012, 237, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Deora, V.; Lee, J.D.; Albornoz, E.A.; McAlary, L.; Jagaraj, C.J.; Robertson, A.A.B.; Atkin, J.D.; Cooper, M.A.; Schroder, K.; Yerbury, J.J.; et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 2020, 68, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burberry, A.; Suzuki, N.; Wang, J.Y.; Moccia, R.; Mordes, D.A.; Stewart, M.H.; Suzuki-Uematsu, S.; Ghosh, S.; Singh, A.; Merkle, F.T.; et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 2016, 8, 347ra93. [Google Scholar] [CrossRef] [Green Version]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Schiffer, D.; Cordera, S.; Cavalla, P.; Migheli, A. Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J. Neurol. Sci. 1996, 139, 27–33. [Google Scholar] [CrossRef]
- Nagy, D.; Kato, T.; Kushner, P.D. Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J. Neurosci. Res. 1994, 38, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Engler, H.; Blomquist, G.; Scott, B.; Wall, A.; Aquilonius, S.M.; Långström, B.; Askmark, H. Evidence for astrocytosis in ALS demonstrated by [11C](l)-deprenyl-D2 PET. J. Neurol. Sci. 2007, 255, 17–22. [Google Scholar] [CrossRef]
- Tong, J.; Huang, C.; Bi, F.; Wu, Q.; Huang, B.; Liu, X.; Li, F.; Zhou, H.; Xia, X.G. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013, 32, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.A.; Garcia, M.L.L.; Mcalonis-downes, M.; Mikse, O.R.R.; Cleveland, D.W.W.; Goldstein, L.S.B.S.B. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.J.; Gendron, T.F.; Grima, J.C.; Sasaguri, H.; Jansen-West, K.; Xu, Y.F.; Katzman, R.B.; Gass, J.; Murray, M.E.; Shinohara, M.; et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 2016, 19, 668–677. [Google Scholar] [CrossRef]
- Wang, L.; Gutmann, D.H.; Roos, R.P. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum. Mol. Genet. 2011, 20, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wang, H.; Qiao, T.; Yang, B.; Aliaga, L.; Qiu, L.; Tan, W.; Salameh, J.; McKenna-Yasek, D.M.; Smith, T.; et al. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, E1121–E1129. [Google Scholar] [CrossRef] [Green Version]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 4697–4702. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Huang, B.; Bi, F.; Yan, L.H.; Tong, J.; Huang, J.; Xia, X.G.; Zhou, H. Profiling the genes affected by pathogenic TDP-43 in astrocytes. J. Neurochem. 2014, 129, 932–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [Green Version]
- Madji Hounoum, B.; Mavel, S.; Coque, E.; Patin, F.; Vourc’h, P.; Marouillat, S.; Nadal-Desbarats, L.; Emond, P.; Corcia, P.; Andres, C.R.; et al. Wildtype motoneurons, ALS-Linked SOD1 mutation and glutamate profoundly modify astrocyte metabolism and lactate shuttling. Glia 2017, 65, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P.L. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 1992, 140, 691–707. [Google Scholar]
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; Fagegaltier, D.; Harris, B.T.; Ostrow, L.W.; et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29, 1164–1177.e5. [Google Scholar] [CrossRef] [Green Version]
- Bonfanti, E.; Bonifacino, T.; Raffaele, S.; Milanese, M.; Morgante, E.; Bonanno, G.; Abbracchio, M.P.; Fumagalli, M. Abnormal Upregulation of GPR17 Receptor Contributes to Oligodendrocyte Dysfunction in SOD1 G93A Mice. Int. J. Mol. Sci. 2020, 21, 2395. [Google Scholar] [CrossRef] [Green Version]
- Molotchnikoff, S.; Cérat, A. Presentation of a remote target influences visual responses of rabbit lateral geniculate cells. Brain Res. Bull. 1990, 24, 381–387. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Meyer, K.; Sherwood, T.W.; Vick, J.; Likhite, S.; Frakes, A.; Miranda, C.J.; Braun, L.; Heath, P.R.; Pineda, R.; et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, E6496–E6505. [Google Scholar] [CrossRef] [Green Version]
- Relaix, F.; Zammit, P.S. Satellite cells are essential for skeletal muscle regeneration: The cell on the edge returns centre stage. Development 2012, 139, 2845–2856. [Google Scholar] [CrossRef] [Green Version]
- Scaramozza, A.; Marchese, V.; Papa, V.; Salaroli, R.; Sorarù, G.; Angelini, C.; Cenacchi, G. Skeletal muscle satellite cells in amyotrophic lateral sclerosis. Ultrastruct. Pathol. 2014, 38, 295–302. [Google Scholar] [CrossRef]
- Manzano, R.; Toivonen, J.M.; Calvo, A.C.; Oliván, S.; Zaragoza, P.; Rodellar, C.; Montarras, D.; Osta, R. Altered in vitro proliferation of mouse SOD1-G93A skeletal muscle satellite cells. Neurodegener. Dis. 2013, 11, 153–164. [Google Scholar] [CrossRef]
- Tokutake, Y.; Yamada, K.; Ohata, M.; Obayashi, Y.; Tsuchiya, M.; Yonekura, S. ALS-linked P56S-VAPB mutation impairs the formation of multinuclear myotube in C2C12 cells. Int. J. Mol. Sci. 2015, 16, 18628–18641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doppler, K.; Mittelbronn, M.; Bornemann, A. Myogenesis in human denervated muscle biopsies. Muscle Nerve 2008, 37, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Pradat, P.F.; Barani, A.; Wanschitz, J.; Dubourg, O.; Lombès, A.; Bigot, A.; Mouly, V.; Bruneteau, G.; Salachas, F.; Lenglet, T.; et al. Abnormalities of satellite cells function in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2011, 12, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Gallo, V.; Vanacore, N.; Bueno-de-Mesquita, H.B.; Vermeulen, R.; Brayne, C.; Pearce, N.; Wark, P.A.; Ward, H.A.; Ferrari, P.; Jenab, M.; et al. Physical activity and risk of Amyotrophic Lateral Sclerosis in a prospective cohort study. Eur. J. Epidemiol. 2016, 31, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Garbugino, L.; Golini, E.; Giuliani, A.; Mandillo, S. Prolonged voluntary running negatively affects survival and disease prognosis of male SOD1G93A low-copy transgenic mice. Front. Behav. Neurosci. 2018, 12, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.; Sinha, K.; Pan, H.; Cui, Y.; Guo, P.; Lin, C.Y.; Yang, F.; Deng, Z.; Eltzschig, H.K.; Lu, A.; et al. Markers of Accelerated Skeletal Muscle Regenerative Response in Murphy Roths Large Mice: Characteristics of Muscle Progenitor Cells and Circulating Factors. Stem Cells 2019, 37, 357–367. [Google Scholar] [CrossRef]
- De Giorgio, F.; Maduro, C.; Fisher, E.M.C.; Acevedo-Arozena, A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. DMM Dis. Model. Mech. 2019, 12, dmm037424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Sharma, K.; Grisotti, G.; Roos, R.P.P. The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiol. Dis. 2009, 35, 234–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditsworth, D.; Maldonado, M.; McAlonis-Downes, M.; Sun, S.; Seelman, A.; Drenner, K.; Arnold, E.; Ling, S.C.; Pizzo, D.; Ravits, J.; et al. Mutant TDP-43 within motor neurons drives disease onset but not progression in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 907–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Grisotti, G.; Roos, R.P. Mutant SOD1 knockdown in all cell types ameliorates disease in G85R SOD1 mice with a limited additional effect over knockdown restricted to motor neurons. J. Neurochem. 2010, 113, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Davis, C.; Sakellariou, G.K.K.; Shi, Y.; Kayani, A.C.C.; Pulliam, D.; Bhattacharya, A.; Richardson, A.; Jackson, M.J.J.; McArdle, A.; et al. CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice. FASEB J. 2013, 27, 3536–3548. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolny, G.; Giacinti, C.; Pelosi, L.; Nicoletti, C.; Winn, N.; Barberi, L.; Molinaro, M.; Rosenthal, N.; Musarò, A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 2005, 168, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrowolny, G.; Bernardini, C.; Martini, M.; Baranzini, M. Muscle Expression of SOD1 G93A Modulates microRNA and mRNA Transcription Pattern Associated with the Myelination Process in the Spinal Cord of Transgenic Mice. Front. Cell. Neurosci. 2015, 9, 463. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolny, G.; Lepore, E.; Martini, M.; Barberi, L.; Nunn, A.; Scicchitano, B.M.; Musarò, A. Metabolic Changes Associated With Muscle Expression of SOD1G93A. Front. Physiol. 2018, 9, 831. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolny, G.; Martini, M.; Scicchitano, B.M.; Romanello, V.; Boncompagni, S.; Nicoletti, C.; Pietrangelo, L.; De Panfilis, S.; Catizone, A.; Bouchè, M.; et al. Muscle Expression of SOD1 G93A Triggers the Dismantlement of Neuromuscular Junction via PKC-Theta. Antioxidants Redox Signal. 2018, 28, 1105–1119. [Google Scholar] [CrossRef]
- Tawara, N.; Yamashita, S.; Kawakami, K.; Kurashige, T.; Zhang, Z.; Tasaki, M.; Yamamoto, Y.; Nishikami, T.; Doki, T.; Zhang, X.; et al. Muscle-dominant wild-type TDP-43 expression induces myopathological changes featuring tubular aggregates and TDP-43-positive inclusions. Exp. Neurol. 2018, 309, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Vogler, T.O.; Wheeler, J.R.; Nguyen, E.D.; Hughes, M.P.; Britson, K.A.; Lester, E.; Rao, B.; Betta, N.D.; Whitney, O.N.; Ewachiw, T.E.; et al. TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 2018, 563, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Sataranatarajan, K.; Qaisar, R.; Davis, C.; Sakellariou, G.K.; Vasilaki, A.; Zhang, Y.; Liu, Y.; Bhaskaran, S.; Mcardle, A.; Jackson, M.; et al. Neuron specific reduction in CuZnSOD is not sufficient to initiate a full sarcopenia phenotype. Redox Biol. 2015, 5, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.H.; Parsadanian, A.S.; Andreeva, A.; Snider, W.D.; Elliott, J.L. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J. Neurosci. 2000, 20, 660–665. [Google Scholar] [CrossRef]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and cellular mechanisms affected in als. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Lepore, E.; Casola, I.; Dobrowolny, G.; Musarò, A. Neuromuscular Junction as an Entity of Nerve-Muscle Communication. Cells 2019, 8, 906. [Google Scholar] [CrossRef] [Green Version]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Rothstein, J.D. From Charcot to Lou Gehrig. Nat. Rev. Neurosci. 2001, 2, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef] [Green Version]
- Boillée, S.; Vande Velde, C.; Cleveland, D.W.W. ALS: A Disease of Motor Neurons and Their Nonneuronal Neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radunović, A.; Mitsumoto, H.; Leigh, P.N. Clinical care of patients with amyotrophic lateral sclerosis. Lancet Neurol. 2007, 6, 913–925. [Google Scholar] [CrossRef]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cachexia and sarcopenia: Mechanisms and potential targets for intervention. Curr. Opin. Pharmacol. 2015, 22, 100–106. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Baeyens, J.P.; Bauer, J.M.; Boirie, Y.; Cederholm, T.; Landi, F.; Martin, F.C.; Michel, J.-P.; Rolland, Y.; Schneider, S.M.; et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 2010, 39, 412–423. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, D.; Ruts, E.; Visser, M.; Heshka, S.; Baumgartner, R.N.; Wang, J.; Pierson, R.N.; Pi-Sunyer, F.X.; Heymsfield, S.B. Weight stability masks sarcopenia in elderly men and women. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E366–E375. [Google Scholar] [CrossRef]
- Lexell, J.; Taylor, C.C.; Sjöström, M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J. Neurol. Sci. 1988, 84, 275–294. [Google Scholar] [CrossRef]
- Klein, C.S.; Marsh, G.D.; Petrella, R.J.; Rice, C.L. Muscle fiber number in the biceps brachii muscle of young and old men. Muscle Nerve 2003, 28, 62–68. [Google Scholar] [CrossRef]
- Bradley, W.G.; Good, P.; Rasool, C.G.; Adelman, L.S. Morphometric and biochemical studies of peripheral nerves in amyotrophic lateral sclerosis. Ann. Neurol. 1983, 14, 267–277. [Google Scholar] [CrossRef]
- Atkin, J.D.; Scott, R.L.; West, J.M.; Lopes, E.; Quah, A.K.J.; Cheema, S.S. Properties of slow- and fast-twitch muscle fibres in a mouse model of amyotrophic lateral sclerosis. Neuromuscul. Disord. 2005, 15, 377–388. [Google Scholar] [CrossRef]
- Frey, D.; Schneider, C.; Xu, L.; Borg, J.; Spooren, W.; Caroni, P. Early and Selective Loss of Neuromuscular Synapse Subtypes with Low Sprouting Competence in Motoneuron Diseases. 2000, 20, 2534–2542. J. Neurosci. 2020, 20, 2534–2542. [Google Scholar] [CrossRef]
- Dengler, R.; Konstanzer, A.; Kuther, G.; Hesse, S.; Wolf, W.; Struppler, A. Amyotrophic lateral sclerosis: Macro-emg and twitch forces of single motor units. Muscle Nerve 1990, 13, 545–550. [Google Scholar] [CrossRef]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Dyck, P.J.; Stevens, J.C.; Mulder, D.W.; Espinosa, R.E. Frequency of nerve fiber degeneration of peripheral motor and sensory neurons in amyotrophic lateral sclerosis. Morphometry of deep and superficial peroneal nerves. Neurology 1975, 25, 781–785. [Google Scholar] [CrossRef]
- Pun, S.; Santos, A.F.; Saxena, S.; Xu, L.; Caroni, P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 2006, 9, 408–419. [Google Scholar] [CrossRef]
- Hegedus, J.; Putman, C.T.; Gordon, T. Time course of preferential motor unit loss in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2007, 28, 154–164. [Google Scholar] [CrossRef]
- Hegedus, J.; Putman, C.T.; Tyreman, N.; Gordon, T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Physiol. 2008, 586, 3337–3351. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Tyreman, N.; Li, S.; Putman, C.T.; Hegedus, J. Neurobiology of Disease Functional over-load saves motor units in the SOD1-G93A transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2010, 37, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Lexell, J. Human aging, muscle mass, and fiber type composition. J. Gerontol. A. Biol. Sci. Med. Sci. 1995, 50, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Tsai, L.K.; Liao, K.Y.; Wu, T.C.; Huang, Y.H.; Huang, Y.C.; Chang, S.W.; Wang, P.Y.; Tsao, Y.P.; Chen, S.L. Muscle-restricted nuclear receptor interaction protein knockout causes motor neuron degeneration through down-regulation of myogenin at the neuromuscular junction. J. Cachexia. Sarcopenia Muscle 2018, 9, 771–785. [Google Scholar] [CrossRef] [PubMed]
- DI Pietro, L.; Baranzini, M.; Berardinelli, M.G.; Lattanzi, W.; Monforte, M.; Tasca, G.; Conte, A.; Logroscino, G.; Michetti, F.; Ricci, E.; et al. Potential therapeutic targets for ALS: MIR206, MIR208b and MIR499 are modulated during disease progression in the skeletal muscle of patients. Sci. Rep. 2017, 7, 9538. [Google Scholar] [CrossRef] [PubMed]
- Veltema, A.N. The case of the saltimbanque prosper lecomte. A contribution to the study of the history of progressive muscular atrophy (aran-duchenne) and amyotrophic lateral sclerosis (charcot). Clin. Neurol. Neurosurg. 1975, 78, 204–209. [Google Scholar] [CrossRef]
- Kuncl, R.W.; Cornblath, D.R.; Griffin, J.W. ASSESSMENT OF THORACIC PARASPINAL MUSCLES IN THE DIAGNOSIS OF ALS. Muscle Nerve 1988, 11, 484–492. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, M.; Pinto, S.; Swash, M. Association of paraspinal and diaphragm denervation in ALS. Amyotroph. Lateral Scler. 2010, 11, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Nozaki, K.; Matsuo, Y.; Tawara, N.; Yamashita, S. Biological significance of target fibres in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1241–1242. [Google Scholar] [CrossRef]
- Schiffman, P.L.; Belsh, M. Pulmonary Function at Diagnosis of Amyotrophic Lateral Sclerosis. Chest 1993, 103, 508–513. [Google Scholar] [CrossRef]
- Achari, A.N.; Anderson, M.S. Serum creatine phospho kinase in amyotrophic lateral sclerosis. Neurology 1974, 24, 477–481. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Sugimoto, H.; Ikeda, K.; Takamiya, K.; Shiojima, T.; Kinoshita, M. Muscle morphometry in amyotrophic lateral sclerosis. Int. J. Neurosci. 1991, 58, 165–170. [Google Scholar] [CrossRef]
- Al-sarraj, S.; King, A.; Cleveland, M.; Pradat, P.; Corse, A.; Rothstein, J.D.; Leigh, P.N.; Abila, B.; Bates, S.; Wurthner, J.; et al. Mitochondrial abnormalities and low grade inflammation are present in the skeletal muscle of a minority of patients with amyotrophic lateral sclerosis; an observational myopathology study. Acta Neuropathol. Commun. 2014, 2, 165. [Google Scholar] [CrossRef] [Green Version]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biedasek, K.; Andres, J.; Mai, K.; Adams, S.; Spuler, S.; Fielitz, J. Skeletal Muscle 11beta-HSD1 Controls Glucocorticoid- Induced Proteolysis and Expression of E3 Ubiquitin Ligases Atrogin-1 and MuRF-1. PLoS ONE 2011, 6, e16674. [Google Scholar] [CrossRef] [PubMed]
- Léger, B.; Vergani, L.; Sorarù, G.; Hespel, P.; Derave, W.; Gobelet, C.; D’Ascenzio, C.; Angelini, C.; Russell, A.P. Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J. 2006, 20, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Ye, F.; Tan, L.; Liu, K.; Xuan, Z.; Zhang, J.; Wang, W.; Zhang, Y.; Jiang, X.; Zhang, D.Y. Alterations of signaling pathways in muscle tissues of patients with amyotrophic lateral sclerosis. Muscle and Nerve 2012, 46, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-induced skeletal myotube hypertrophy by Pl(3)K/Alt/mTOR and Pl(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Lunetta, C.; Serafini, M.; Prelle, A.; Magni, P.; Dozio, E.; Ruscica, M.; Sassone, J.; Colciago, C.; Moggio, M.; Corbo, M.; et al. Impaired expression of insulin-like growth factor-1 system in skeletal muscle of amyotrophic lateral sclerosis patients. Muscle and Nerve 2012, 45, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolny, G.; Aucello, M.; Musarò, A. Muscle atrophy induced by SOD1G93A expression does not involve the activation of caspase in the absence of denervation. Skelet. Muscle 2011, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Jagoe, R.T.; Goldberg, A.L. What do we really know about the ubiquitin ± proteasome pathway in muscle atrophy ? Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 183–190. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2019, 9, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Gill, C.; Phelan, J.P.; Theo, H.; Kidd, J.D.; Tassinari, V.R.; Levine, B.; Wang, M.Z.; Moreno, A.; Thompson, K.; Maier, M.; et al. SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS. Sci. Rep. 2019, 9, 6724. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Baldie, G.; Periz, G.; Wang, J. RNA-Processing Protein TDP-43 Regulates FOXO-Dependent Protein Quality Control in Stress Response. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef]
- Picchiarelli, G.; Dupuis, L. Role of RNA Binding Proteins with prion-like domains in muscle and neuromuscular diseases. Cell Stress 2020, 4, 76–91. [Google Scholar] [CrossRef] [Green Version]
- Dejesus-hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Teyssou, E.; Chartier, L.; Amador, M.; Lam, R.; Lautrette, G.; Nicol, M.; Machat, S.; Da, S.; Moigneu, C.; Mairey, M.; et al. Neurobiology of Aging Novel UBQLN2 mutations linked to amyotrophic lateral sclerosis and atypical hereditary spastic paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiol. Aging 2017, 58, 239.e11–239.e20. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Report Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 58, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Saeki, Y. JB Special Review — Recent Topics in Ubiquitin-Proteasome System and Autophagy Ubiquitin recognition by the proteasome. J. Biochem. 2017, 161, 113–124. [Google Scholar] [CrossRef]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62 / SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Anagnostou, G.; Chai, A.; Withers, J.; Morris, A.; Adhikaree, J.; Pennetta, G.; Belleroche, J.S. De Characterization of the Properties of a Novel Mutation in VAPB in Familial Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2010, 285, 40266–40281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Lan, M.; Mojsilovic-petrovic, J.; Choi, W.H.; Safren, N.; Barmada, S.; Lee, M.J.; Kalb, R. The Proline / Arginine Dipeptide from Hexanucleotide Repeat Expanded C9ORF72 Inhibits the Proteasome. eNeuro 2017. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.; Liao, Y.; Chen, P.; Guo, Y.; Chen, Y.; Jih, K.; Lin, K.; Soong, B.; Tsai, C. Neurobiology of Aging Investigating CCNF mutations in a Taiwanese cohort with amyotrophic lateral sclerosis. Neurobiol. Aging 2017, 243.e1–243.e6. [Google Scholar] [CrossRef]
- Türk, M.; Haaker, G.; Winter, L.; Just, W.; Nickel, F.T.; Linker, R.A.; Chevessier, F.; Schröder, R. C9ORF72-ALS: P62- and ubiquitin-aggregation pathology in skeletal muscle. Muscle Nerve 2014, 50, 454–455. [Google Scholar] [CrossRef] [PubMed]
- Leigh, P.N.; Whitwell, H.; Garofalo, O.; Buller, J.; Swash, M.; Martin, J.E.; Gallo, J.M.; Weller, R.O.; Anderton, B.H. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 1991, 114, 775–788. [Google Scholar] [CrossRef]
- Shibata, N.; Hirano, A.; Kobayashi, M.; Sasaki, S.; Kato, T.; Matsumoto, S.; Shiozawa, Z.; Komori, T.; Ikemoto, A.; Umahara, T. Cu/Zn superoxide dismutase-like immunoreactivity in Lewy body-like inclusions of sporadic amyotrophic lateral sclerosis. Neurosci. Lett. 1994, 179, 149–152. [Google Scholar] [CrossRef]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Knebel, A.; Marchesi, F.; Kurz, T.; Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome Article UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Cykowski, M.D.; Dickson, D.W.; Powell, S.Z.; Arumanayagam, A.S.; Rivera, A.L.; Appel, S.H. Dipeptide repeat ( DPR ) pathology in the skeletal muscle of ALS patients with C9ORF72 repeat expansion. Acta Neuropathol. 2019, 138, 667–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabashi, E.; Agar, J.N.; Taylor, D.M.; Minotti, S.; Durham, H.D. Focal dysfunction of the proteasome: A pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2004, 89, 1325–1335. [Google Scholar] [CrossRef]
- Cicardi, M.E.; Marrone, L.; Azzouz, M.; Trotti, D. Proteostatic imbalance and protein spreading in amyotrophic lateral sclerosis. EMBO J. 2021, 40, e106389. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Frantz, J.D.; Tawa, N.E.; Melendez, P.A.; Oh, B.C.; Lidov, H.G.W.; Hasselgren, P.O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackman, R.W.; Kandarian, S.C. The molecular basis of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 2004, 287, 834–843. [Google Scholar] [CrossRef] [Green Version]
- Halter, B.; Gonzalez de Aguilar, J.L.; Rene, F.; Petri, S.; Fricker, B.; Echaniz-Laguna, A.; Dupuis, L.; Larmet, Y.; Loeffler, J.P. Oxidative stress in skeletal muscle stimulates early expression of Rad in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2010, 48, 915–923. [Google Scholar] [CrossRef]
- De Aguilar, J.L.G.D.L.G.; Echaniz-Laguna, A.; Fergani, A.; René, F.; Meininger, V.; Loeffler, J.P.P.; Dupuis, L. Amyotrophic lateral sclerosis: All roads lead to Rome. J. Neurochem. 2007, 101, 1153–1160. [Google Scholar] [CrossRef]
- Ash, P.E.A.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.; Dejesus-hernandez, M.; Van Blitterswijk, M.M.; Jansen-west, K.; Paul, J.W., 3rd; Rademakers, R.; et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Schludi, M.H.; May, S.; Grässer, F.A.; Rentzsch, K.; Kremmer, E.; Küpper, C.; Klopstock, T.; Diehl-schmid, J.; Fassbender, K.; Förstl, H.; et al. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 2015, 130, 537–555. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.P.; Higginbottom, A.; Mcgown, A.; Castelli, L.M.; James, E.; Hautbergue, G.M.; Shaw, P.J.; Ramesh, T.M. Stable transgenic C9orf72 zebrafish model key aspects of the ALS / FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 2018, 6, 125. [Google Scholar] [CrossRef] [Green Version]
- Freibaum, B.D.; Lu, Y.; Lopez-gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef]
- Cykowski, M.D.; Powell, S.Z.; Appel, J.W.; Arumanayagam, A.S.; Rivera, A.L.; Appel, S.H. Phosphorylated TDP-43 (pTDP-43) aggregates in the axial skeletal muscle of patients with sporadic and familial amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2018, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Huisman, M.H.B.; Seelen, M.; De Jong, S.W.; Dorresteijn, K.R.I.S.; Van Doormaal, P.T.C.; Van Der Kooi, A.J.; De Visser, M.; Schelhaas, H.J.; Van Den Berg, L.H.; Veldink, J.H. Lifetime physical activity and the risk of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Kihira, T.; Kondo, T.; Kobashi, G.; Washio, M.; Sasaki, S.; Yokoyama, T.; Miyake, Y.; Sakamoto, N.; Inaba, Y.; et al. Lifestyle Factors and Risk of Amyotrophic Lateral Sclerosis: A Case-Control Study in Japan. Ann. Epidemiol. 2009, 19, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Visser, A.E.; Rooney, J.P.K.; D’ovidio, F.; Westeneng, H.J.; Vermeulen, R.C.H.; Beghi, E.; Chiò, A.; Logroscino, G.; Bru-No, A.; Fanti, C.; et al. Multicentre, cross-cultural, population-based, case-control study of physical activity as risk factor for amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Hållmarker, U.; James, S.; Ingre, C.; Michaëlsson, K.; Ahlbom, A.; Feychting, M. Amyotrophic lateral sclerosis among cross-country skiers in Sweden. Eur. J. Epidemiol. 2016, 31, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Gotkine, M.; Friedlander, Y.; Hochner, H. Triathletes are over-represented in a population of patients with ALS. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 534–536. [Google Scholar] [CrossRef]
- Pupillo, E.; Bianchi, E.; Vanacore, N.; Montalto, C.; Ricca, G.; Robustelli Della Cuna, F.S.; Fumagalli, F.; Castellani, M.; Poli, F.; Romeo, F.; et al. Increased risk and early onset of ALS in professional players from Italian Soccer Teams. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 403–409. [Google Scholar] [CrossRef]
- Lacorte, E.; Ferrigno, L.; Leoncini, E.; Corbo, M.; Boccia, S.; Vanacore, N. Physical activity, and physical activity related to sports, leisure and occupational activity as risk factors for ALS: A systematic review. Neurosci. Biobehav. Rev. 2016, 66, 61–79. [Google Scholar] [CrossRef]
- Harwood, C.A.; Westgate, K.; Gunstone, S.; Brage, S.; Wareham, N.J.; McDermott, C.J.; Shaw, P.J. Long-term physical activity: An exogenous risk factor for sporadic amyotrophic lateral sclerosis? Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Li, X.; Li, C.; Tsang, R.C.C.; Chen, Y.; Ge, Y.; Gao, Q. Effects of Exercise in Patients With Amyotrophic Lateral Sclerosis. Am. J. Phys. Med. Rehabil. 2020, 99, 801–810. [Google Scholar] [CrossRef]
- Julian, T.H.; Glascow, N.; Dylan Fisher Barry, A.; Moll, T.; Harvey, C.; Klimentidis, Y.C.; Newell, M.; Zhang, S.; Snyder, M.P.; Cooper-Knock, J.; et al. Physical exercise is a risk factor for amyotrophic lateral sclerosis: Convergent evidence from mendelian randomisation, transcriptomics and risk genotypes. EBioMedicine 2021, 69, 103438. [Google Scholar]
- Liu, G.; Ou, S.; Cui, H.; Li, X.; Yin, Z.; Gu, D.; Wang, Z. Head Injury and Amyotrophic Lateral Sclerosis: A Meta-Analysis. Neuroepidemiology 2021, 55, 11–19. [Google Scholar] [CrossRef]
- Chen, H.; Richard, M.; Sandler, D.P.; Umbach, D.M.; Kamel, F. Head injury and amyotrophic lateral sclerosis. Am. J. Epidemiol. 2007, 166, 810–816. [Google Scholar] [CrossRef] [Green Version]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef] [Green Version]
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor neuron susceptibility in ALS/FTD. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, S.; Caroni, P. Selective Neuronal Vulnerability in Neurodegenerative Diseases: From Stressor Thresholds to Degeneration. Neuron 2011, 71, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Kasarskis, E.J.; Berryman, S.; Vanderleest, J.G.; Schneider, A.R.; McClain, C.J. Nutritional status of patients with amyotrophic lateral sclerosis: Relation to the proximity of death. Am. J. Clin. Nutr. 1996, 63, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouteloup, C.; Desport, J.-C.; Clavelou, P.; Guy, N.; Derumeaux-Burel, H.; Ferrier, A.; Couratier, P. Hypermetabolism in ALS patients: An early and persistent phenomenon. J. Neurol. 2009, 256, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Desport, J.C.; Preux, P.M.; Magy, L.; Boirie, Y.; Vallat, J.M.; Couratier, P. Factors correlated with hypermetabolism in patients with ALS. Am. J. Clin. Nutr. 2001, 74, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Steyn, F.J.; Ioannides, Z.A.; van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; van den Berg, L.H.; et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jésus, P.; Fayemendy, P.; Nicol, M.; Lautrette, G.; Sourisseau, H.; Preux, P.-M.; Desport, J.-C.; Marin, B.; Couratier, P. Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2018, 25, 97–104. [Google Scholar] [CrossRef]
- Abdullahi, A.; Jeschke, M.G. White Adipose Tissue Browning: A Double-edged Sword. Trends Endocrinol. Metab. 2016, 27, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.-L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.-J.; Lacomblez, L.; Loeffler, J.-P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Nodera, H.; Takamatsu, N.; Muguruma, N.; Ukimoto, K.; Nishio, S.; Oda, M.; Izumi, Y.; Kaji, R.; Hiroyuki Nodera, C. Frequent hepatic steatosis in amyotrophic lateral sclerosis: Implication for systemic involvement. Neurol. Clin. Neurosci. 2015, 3, 58–62. [Google Scholar] [CrossRef]
- Huang, R.; Guo, X.; Chen, X.; Zheng, Z.; Wei, Q.; Cao, B.; Zeng, Y.; Shang, H. The serum lipid profiles of amyotrophic lateral sclerosis patients: A study from south-west China and a meta-analysis. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 359–365. [Google Scholar] [CrossRef]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.-L.A.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.-P.J.; et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Giacovazzo, G.; Loeffler, J.P.; Renè, F.; Rosina, M.; Quessada, C.; Proietti, D.; Heil, C.; Rossi, S.; et al. Skeletal-Muscle Metabolic Reprogramming in ALS-SOD1G93A Mice Predates Disease Onset and Is A Promising Therapeutic Target. iScience 2020, 23, 101087. [Google Scholar] [CrossRef]
- Camerino, G.M.; Fonzino, A.; Conte, E.; De Bellis, M.; Mele, A.; Liantonio, A.; Tricarico, D.; Tarantino, N.; Dobrowolny, G.; Musarò, A.; et al. Elucidating the Contribution of Skeletal Muscle Ion Channels to Amyotrophic Lateral Sclerosis in search of new therapeutic options. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bittel, D.C.; Jaiswal, J.K. Contribution of extracellular vesicles in rebuilding injured muscles. Front. Physiol. 2019, 10, 828. [Google Scholar] [CrossRef] [Green Version]
- Baci, D.; Chirivì, M.; Pace, V.; Maiullari, F.; Milan, M.; Rampin, A.; Somma, P.; Presutti, D.; Garavelli, S.; Bruno, A.; et al. Extracellular Vesicles from Skeletal Muscle Cells Efficiently Promote Myogenesis in Induced Pluripotent Stem Cells. Cells 2020, 9, 1527. [Google Scholar] [CrossRef]
- Trovato, E.; Di Felice, V.; Barone, R. Extracellular vesicles: Delivery vehicles of myokines. Front. Physiol. 2019, 10, 522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.T.; King, K.Y.; Brett, J.O.; Cromie, M.J.; Charville, G.W.; Maguire, K.K.; Brunson, C.; Mastey, N.; Liu, L.; Tsai, C.R.; et al. MTORC1 controls the adaptive transition of quiescent stem cells from G 0 to GAlert. Nature 2014, 510, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Schroeder, M.D.; Ma, C.; Rando, T.A. HGFA Is an Injury-Regulated Systemic Factor that Induces the Transition of Stem Cells into GAlert. Cell Rep. 2017, 19, 479–486. [Google Scholar] [CrossRef]
- Anderson, J.E.; Do, M.K.Q.; Daneshvar, N.; Suzuki, T.; Dort, J.; Mizunoya, W.; Tatsumi, R. The role of semaphorin3A in myogenic regeneration and the formation of functional neuromuscular junctions on new fibres. Biol. Rev. 2017, 92, 1389–1405. [Google Scholar] [CrossRef]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Di Scala, F.; Rene, F.; De Tapia, M.; Pradat, P.F.; Lacomblez, L.; Seihlan, D.; Prinjha, R.; Walsh, F.S.; et al. Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis. Neurobiol. Dis. 2002, 10, 358–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gasperi, R.; Hamidi, S.; Harlow, L.M.; Ksiezak-Reding, H.; Bauman, W.A.; Cardozo, C.P. Denervation-related alterations and biological activity of miRNAs contained in exosomes released by skeletal muscle fibers. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Ditlevsen, D.K.; Povlsen, G.K.; Berezin, V.; Bock, E. NCAM-induced intracellular signaling revisited. J. Neurosci. Res. 2008, 86, 727–743. [Google Scholar] [CrossRef]
- Schwab, M.E. Functions of Nogo proteins and their receptors in the nervous system. Nat. Rev. Neurosci. 2010, 11, 799–811. [Google Scholar] [CrossRef]
- Teng, F.Y.H.; Tang, B.L. Nogo-A and Nogo-66 receptor in amyotrophic lateral sclerosis. J. Cell. Mol. Med. 2008, 12, 1199–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meininger, V.; Genge, A.; van den Berg, L.H.; Robberecht, W.; Ludolph, A.; Chio, A.; Kim, S.H.; Leigh, P.N.; Kiernan, M.C.; Shefner, J.M.; et al. Safety and efficacy of ozanezumab in patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2017, 16, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Park, J.H.; Kang, U.B.; Choi, S.K.; Elfadl, A.; Ullah, H.M.A.; Chung, M.J.; Son, J.Y.; Yun, H.H.; Park, J.M.; et al. Nogo-A regulates myogenesis via interacting with Filamin-C. Cell Death Discov. 2021, 7, 1. [Google Scholar] [CrossRef]
- Covington, J.D.; Tam, C.S.; Bajpeyi, S.; Galgani, J.E.; Noland, R.C.; Smith, S.R.; Redman, L.M.; Ravussin, E. Myokine Expression in Muscle and Myotubes in Response to Exercise Stimulation. Med. Sci. Sports Exerc. 2016, 48, 384–390. [Google Scholar] [CrossRef] [Green Version]
- Delezie, J.; Weihrauch, M.; Maier, G.; Tejero, R.; Ham, D.J.; Gill, J.F.; Karrer-Cardel, B.; Rüegg, M.A.; Tabares, L.; Handschin, C. BDNF is a mediator of glycolytic fiber-type specification in mouse skeletal muscle. Proc. Natl. Acad. Sci. USA 2019, 116, 16111–16120. [Google Scholar] [CrossRef] [Green Version]
- Gerenu, G.; Martisova, E.; Ferrero, H.; Carracedo, M.; Rantamäki, T.; Ramirez, M.J.; Gil-Bea, F.J. Modulation of BDNF cleavage by plasminogen-activator inhibitor-1 contributes to Alzheimer’s neuropathology and cognitive deficits. Biochim. Biophys. Acta - Mol. Basis Dis. 2017, 1863, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.M.W.; Pedersen, B.K. The anti-inflammatory effect of exercise. J. Appl. Physiol. 2005, 98, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Lehrskov, L.L.; Christensen, R.H. The role of interleukin-6 in glucose homeostasis and lipid metabolism. Semin. Immunopathol. 2019, 41, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Higashimori, T.; Park, S.Y.; Choi, H.; Dong, J.; Kim, Y.J.; Noh, H.L.; Cho, Y.R.; Cline, G.; Kim, Y.B.; et al. Differential Effects of Interleukin-6 and -10 on Skeletal Muscle and Liver Insulin Action In Vivo. Diabetes 2004, 53, 1060–1067. [Google Scholar] [CrossRef] [Green Version]
- Leibinger, M.; Müller, A.; Gobrecht, P.; Diekmann, H.; Andreadaki, A.; Fischer, D. Interleukin-6 contributes to CNS axon regeneration upon inflammatory stimulation. Cell Death Dis. 2013, 4, e609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibinger, M.; Zeitler, C.; Gobrecht, P.; Andreadaki, A.; Gisselmann, G.; Fischer, D. Transneuronal delivery of hyper-interleukin-6 enables functional recovery after severe spinal cord injury in mice. Nat. Commun. 2021, 12, 391. [Google Scholar] [CrossRef]
- Hu, Y.; Cao, C.; Qin, X.Y.; Yu, Y.; Yuan, J.; Zhao, Y.; Cheng, Y. Increased peripheral blood inflammatory cytokine levels in amyotrophic lateral sclerosis: A meta-analysis study. Sci. Rep. 2017, 7, 9094. [Google Scholar] [CrossRef]
- Han, Y.; Ripley, B.; Serada, S.; Naka, T.; Fujimoto, M. Interleukin-6 Deficiency Does Not Affect Motor Neuron Disease Caused by Superoxide Dismutase 1 Mutation. PLoS ONE 2016, 11, e0153399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyileten, C.; Sharif, L.; Wicik, Z.; Jakubik, D.; Jarosz-Popek, J.; Soplinska, A.; Postula, M.; Czlonkowska, A.; Kaplon-Cieslicka, A.; Mirowska-Guzel, D. The Relation of the Brain-Derived Neurotrophic Factor with MicroRNAs in Neurodegenerative Diseases and Ischemic Stroke. Mol. Neurobiol. 2021, 58, 329–347. [Google Scholar] [CrossRef] [PubMed]
- Halievski, K.; Xu, Y.; Haddad, Y.W.; Tang, Y.P.; Yamada, S.; Katsuno, M.; Adachi, H.; Sobue, G.; Breedlove, S.M.; Jordan, C.L. Muscle BDNF improves synaptic and contractile muscle strength in Kennedy’s disease mice in a muscle-type specific manner. J. Physiol. 2020, 598, 2719–2739. [Google Scholar] [CrossRef]
- Mousavi, K.; Jasmin, B.J. BDNF is expressed in skeletal muscle satellite cells and inhibits myogenic differentiation. J. Neurosci. 2006, 26, 5739–5749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouel, F.; Rolland, A.S.; Devedjian, J.C.; Burnouf, T.; Devos, D. Past and future of neurotrophic growth factors therapies in ALS: From single neurotrophic growth factor to stem cells and human platelet lysates. Front. Neurol. 2019, 10, 835. [Google Scholar] [CrossRef]
- Le Gall, L.; Duddy, W.J.; Martinat, C.; Mariot, V.; Connolly, O.; Milla, V.; Anakor, E.; Ouandaogo, Z.G.; Millecamps, S.; Lainé, J.; et al. Muscle cells of sporadic ALS patients secrete neurotoxic vesicles. medRxiv 2021. [Google Scholar] [CrossRef]
- Picchiarelli, G.; Demestre, M.; Zuko, A.; Been, M.; Higelin, J.; Dieterlé, S.; Goy, M.A.; Mallik, M.; Sellier, C.; Scekic-Zahirovic, J.; et al. FUS-mediated regulation of acetylcholine receptor transcription at neuromuscular junctions is compromised in amyotrophic lateral sclerosis. Nat. Neurosci. 2019, 22, 1793–1805. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.P.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet. Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.; Arzberger, T.; Kremmer, E.; Troost, D.; Lorenzl, S.; Mori, K.; Weng, S.-M.; Haass, C.; Kretzschmar, H.A.; Edbauer, D.; et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 2013, 126, 859–879. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.-J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- May, S.; Hornburg, D.; Schludi, M.H.; Arzberger, T.; Rentzsch, K.; Schwenk, B.M.; Grässer, F.A.; Mori, K.; Kremmer, E.; Banzhaf-Strathmann, J.; et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014, 128, 485–503. [Google Scholar] [CrossRef] [Green Version]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef] [Green Version]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, J.; Gendron, T.F.; Prudencio, M.; Sasaguri, H.; Zhang, Y.-J.; Castanedes-Casey, M.; Lee, C.W.; Jansen-West, K.; Kurti, A.; Murray, M.E.; et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 2015, 348, 1151–1154. [Google Scholar] [CrossRef] [Green Version]
- Stopford, M.J.; Higginbottom, A.; Hautbergue, G.M.; Cooper-Knock, J.; Mulcahy, P.J.; De Vos, K.J.; Renton, A.E.; Pliner, H.; Calvo, A.; Chio, A.; et al. C9ORF72 hexanucleotide repeat exerts toxicity in a stable, inducible motor neuronal cell model, which is rescued by partial depletion of Pten. Hum. Mol. Genet. 2017, 26, 1133–1145. [Google Scholar] [CrossRef]
- Swaminathan, A.; Bouffard, M.; Liao, M.; Ryan, S.; Callister, J.B.; Pickering-Brown, S.M.; Armstrong, G.A.B.; Drapeau, P. Expression of C9orf72-related dipeptides impairs motor function in a vertebrate model. Hum. Mol. Genet. 2018, 27, 1754–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-J.; Gendron, T.F.; Ebbert, M.T.W.; O’Raw, A.D.; Yue, M.; Jansen-West, K.; Zhang, X.; Prudencio, M.; Chew, J.; Cook, C.N.; et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 2018, 24, 1136–1142. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Guo, L.; Gonzales, P.K.; Gendron, T.F.; Wu, Y.; Jansen-West, K.; O’Raw, A.D.; Pickles, S.R.; Prudencio, M.; Carlomagno, Y.; et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 2019, 363, eaav2606. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Munoz, D.G.; Greene, C.; Perl, D.P.; Selkoe, D.J. Accumulation of phosphorylated neurofilaments in anterior horn motoneurons of amyotrophic lateral sclerosis patients. J. Neuropathol. Exp. Neurol. 1988, 47, 9–18. [Google Scholar] [CrossRef]
- Nakamura-Shindo, K.; Sakai, K.; Shimizu, A.; Ishida, C.; Yamada, M. Accumulation of phosphorylated TDP-43 in the cytoplasm of Schwann cells in a case of sporadic amyotrophic lateral sclerosis. Neuropathology 2020, 40, 606–610. [Google Scholar] [CrossRef]
- Ghasemi, M.; Brown, R.H. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Montiel, M.J.; Chaineau, M.; Durcan, T.M. The Neglected Genes of ALS: Cytoskeletal Dynamics Impact Synaptic Degeneration in ALS. Front. Cell. Neurosci. 2020, 14, 380. [Google Scholar] [CrossRef]
- Campbell, P.D.; Shen, K.; Sapio, M.R.; Glenn, T.D.; Talbot, W.S.; Marlow, F.L. Unique function of Kinesin Kif5A in localization of mitochondria in axons. J. Neurosci. 2014, 34, 14717–14732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, C.H.; Roberts, E.A.; Her, L.S.; Liu, X.; Williams, D.S.; Cleveland, D.W.; Goldstein, L.S.B. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J. Cell Biol. 2003, 161, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Yilmaz, R.; Müller, K.; Grehl, T.; Petri, S.; Meyer, T.; Grosskreutz, J.; Weydt, P.; Ruf, W.; Neuwirth, C.; et al. Hot-spot KIF5A mutations cause familial ALS. Brain 2018, 141, 688–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konno, T.; Ross, O.A.; Teive, H.A.G.; Sławek, J.; Dickson, D.W.; Wszolek, Z.K. DCTN1-related neurodegeneration: Perry syndrome and beyond. Park. Relat. Disord. 2017, 41, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, K.I.; Saiki, S.; Furuya, N.; Imamichi, Y.; Tsuboi, Y.; Hattori, N. p150 glued deficiency impairs effective fusion between autophagosomes and lysosomes due to their redistribution to the cell periphery. Neurosci. Lett. 2019, 690, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.F.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.R.; Sumner, C.J.; Caviston, J.P.; Tokito, M.K.; Ranganathan, S.; Ligon, L.A.; Wallace, K.E.; LaMonte, B.H.; Harmison, G.G.; Puls, I.; et al. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J. Cell Biol. 2006, 172, 733–745. [Google Scholar] [CrossRef]
- Münch, C.; Sedlmeier, R.; Meyer, T.; Homberg, V.; Sperfeld, A.D.; Kurt, A.; Prudlo, J.; Peraus, G.; Hanemann, C.O.; Stumm, G.; et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene ALS. Neurology 2004, 63, 724–726. [Google Scholar] [CrossRef]
- Leung, C.L.; He, C.Z.; Kaufmann, P.; Chin, S.S.; Naini, A.; Liem, R.K.H.; Mitsumoto, H.; Hays, A.P. A pathogenic peripherin gene mutation in a patient with amyotrophic lateral sclerosis. Brain Pathol. 2004, 14, 290–296. [Google Scholar] [CrossRef]
- Eriksson, K.S.; Zhang, S.; Lin, L.; Larivière, R.C.; Julien, J.P.; Mignot, E. The type III neurofilament peripherin is expressed in the tuberomammillary neurons of the mouse. BMC Neurosci. 2008, 9, 26. [Google Scholar] [CrossRef] [Green Version]
- Troy, C.M.; Muma, N.A.; Greene, L.A.; Price, D.L.; Shelanski, M.L. Regulation of peripherin and neurofilament expression in regenerating rat motor neurons. Brain Res. 1990, 529, 232–238. [Google Scholar] [CrossRef]
- Figlewicz, D.A.; Krizus, A.; Martinoli, M.G.; Meininger, V.; Dib, M.; Rouleau, G.A.; Julien, J.P. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet. 1994, 3, 1757–1761. [Google Scholar] [CrossRef]
- Xiao, S.; McLean, J.; Robertson, J. Neuronal intermediate filaments and ALS: A new look at an old question. Biochim. Biophys. Acta. Mol. Basis Dis. 2006, 1762, 1001–1012. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.N.; Ticozzi, N.; Fallini, C.; Gkazi, A.S.; Topp, S.; Kenna, K.P.; Scotter, E.L.; Kost, J.; Keagle, P.; Miller, J.W.; et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 2014, 84, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Warita, H.; Itoyama, Y.; Abe, K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1999, 819, 120–131. [Google Scholar] [CrossRef]
- Bilsland, L.G.; Sahai, E.; Kelly, G.; Golding, M.; Greensmith, L.; Schiavo, G. Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. USA. 2010, 107, 20523–20528. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Shanware, N.P.; Bowler, M.J.; Tibbetts, R.S. Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J. Biol. Chem. 2010, 285, 34097–34105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Berghe, P.V.; Timmerman, V.; Robberecht, W.; Van Den Bosch, L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Burk, K.; Pasterkamp, R.J. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 2019, 137, 859–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoell, J.I.; Larsson, E.; Runge, S.; Nusbaum, J.D.; Duggimpudi, S.; Farazi, T.A.; Hafner, M.; Borkhardt, A.; Sander, C.; Tuschl, T. RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol. 2011, 18, 1428–1431. [Google Scholar] [CrossRef] [PubMed]
- Orozco, D.; Edbauer, D. FUS-mediated alternative splicing in the nervous system: Consequences for ALS and FTLD. J. Mol. Med. 2013, 91, 1343–1354. [Google Scholar] [CrossRef]
- Kanouchi, T.; Ohkubo, T.; Yokota, T. Can regional spreading of amyotrophic lateral sclerosis motor symptoms be explained by prion-like propagation? J. Neurol. Neurosurg. Psychiatry 2012, 83, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Jo, M.; Lee, S.; Jeon, Y.M.; Kim, S.; Kwon, Y.; Kim, H.J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef]
- Prusiner, S.B. Shattuck lecture--neurodegenerative diseases and prions. N. Engl. J. Med. 2001, 344, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell. Neurosci. 2019, 100, 103396. [Google Scholar] [CrossRef]
- Wang, Y.-T.; Kuo, P.-H.; Chiang, C.-H.; Liang, J.-R.; Chen, Y.-R.; Wang, S.; Shen, J.C.K.; Yuan, H.S. The truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. J. Biol. Chem. 2013, 288, 9049–9057. [Google Scholar] [CrossRef] [Green Version]
- Münch, C.; Bertolotti, A. Exposure of hydrophobic surfaces initiates aggregation of diverse ALS-causing superoxide dismutase-1 mutants. J. Mol. Biol. 2010, 399, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Durazo, A.; Sohn, S.H.; Strong, C.D.; Gralla, E.B.; Whitelegge, J.P.; Valentine, J.S. Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc. Natl. Acad. Sci. USA 2008, 105, 18663–18668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.K.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.S.; Michel, C.H.; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef] [Green Version]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef] [Green Version]
- Dormann, D.; Haass, C. TDP-43 and FUS: A nuclear affair. Trends Neurosci. 2011, 34, 339–348. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Van den Bos, M.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int. J. Mol. Sci. 2019, 20, 2818. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Chakkalakal, J.V. The Composition, Development, and Regeneration of Neuromuscular Junctions, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; Volume 126. [Google Scholar]
- Fuertes-Alvarez, S.; Izeta, A. Terminal Schwann Cell Aging: Implications for Age-Associated Neuromuscular Dysfunction. Aging Dis. 2021, 12, 494. [Google Scholar] [CrossRef]
- Martineau, É.; Arbour, D.; Vallée, J.; Robitaille, R. Properties of glial cell at the neuromuscular junction are incompatible with synaptic repair in the SOD1G37R ALS mouse model. J. Neurosci. 2020, 40, 7759–7777. [Google Scholar] [CrossRef]
- Barik, A.; Li, L.; Sathyamurthy, A.; Xiong, W.C.; Mei, L.; Barik, X.A.; Li, X.L.; Sathyamurthy, X.A.; Xiong, X.W.; Mei, L. Schwann cells in neuromuscular junction formation and maintenance. J. Neurosci. 2016, 36, 9770–9781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Tian, L.; Mikesh, M.; Lichtman, J.W.; Thompson, W.J. Terminal schwann cells participate in neuromuscular synapse remodeling during reinnervation following nerve injury. J. Neurosci. 2014, 34, 6323–6333. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, M.E.; Meehan, G.R.; Robinson, S.; Yao, D.; McGonigal, R.; Willison, H.J. Perisynaptic Schwann cells phagocytose nerve terminal debris in a mouse model of Guillain-Barré syndrome. J. Peripher. Nerv. Syst. 2020, 25, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Court, F.A.; Gillingwater, T.H.; Melrose, S.; Sherman, D.L.; Greenshields, K.N.; Morton, A.J.; Harris, J.B.; Willison, H.J.; Ribchester, R.R. Identity, developmental restriction and reactivity of extralaminar cells capping mammalian neuromuscular junctions. J. Cell Sci. 2008, 121, 3901–3911. [Google Scholar] [CrossRef] [Green Version]
- Mathis, S.; Couratier, P.; Julian, A.; Vallat, J.M.; Corcia, P.; Le Masson, G. Management and therapeutic perspectives in amyotrophic lateral sclerosis. Expert Rev. Neurother. 2017, 17, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Gotaas, H.T.; Skeie, G.O.; Gilhus, N.E. Myasthenia gravis and amyotrophic lateral sclerosis: A pathogenic overlap. Neuromuscul. Disord. 2016, 26, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Martineau, É.; Di Polo, A.; Velde, C.V.; Robitaille, R. Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. Elife 2018, 7, e41973. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016, 76, 35–47. [Google Scholar] [CrossRef]
- Rocha, M.C.; Pousinha, P.A.; Correia, A.M.; Sebastião, A.M.; Ribeiro, J.A. Early Changes of Neuromuscular Transmission in the SOD1(G93A) Mice Model of ALS Start Long before Motor Symptoms Onset. PLoS ONE 2013, 8, e73846. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, E.; Martineau, É.; Robitaille, R. Opposite synaptic alterations at the neuromuscular junction in an ALS mouse model: When motor units matter. J. Neurosci. 2017, 37, 8901–8918. [Google Scholar] [CrossRef]
- Arbour, D.; Tremblay, E.; Martineau, É.; Julien, J.P.; Robitaille, R. Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J. Neurosci. 2015, 35, 688–706. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Goldberg, A.L.L. SIRT1 Protein, by Blocking the Activities of Transcription Factors FoxO1 and FoxO3, Inhibits Muscle Atrophy and Promotes Muscle Growth. J. Biol. Chem. 2013, 288, 30515–30526. [Google Scholar] [CrossRef] [Green Version]
- Harrison, J.M.; Rafuse, V.F. Muscle fiber-type specific terminal Schwann cell pathology leads to sprouting deficits following partial denervation in SOD1G93A mice. Neurobiol. Dis. 2020, 145, 105052. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.I.; Bahr, B.A.; Seburn, K.L.; Pinter, M.J. Abnormal response of distal Schwann cells to denervation in a mouse model of motor neuron disease. Exp Neurol 2016, 278, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappello, V.; Francolini, M. Neuromuscular junction dismantling in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2017, 18, 2092. [Google Scholar] [CrossRef] [PubMed]
- Santosa, K.B.; Keane, A.M.; Jablonka-Shariff, A.; Vannucci, B.; Snyder-Warwick, A.K. Clinical relevance of terminal Schwann cells: An overlooked component of the neuromuscular junction. J. Neurosci. Res. 2018, 96, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Bruneteau, G.; Bauché, S.; Gonzalez de Aguilar, J.L.; Brochier, G.; Mandjee, N.; Tanguy, M.-L.; Hussain, G.; Behin, A.; Khiami, F.; Sariali, E.; et al. Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann. Clin. Transl. Neurol. 2015, 2, 362–372. [Google Scholar] [CrossRef]
- Darabid, H.; Perez-Gonzalez, A.P.; Robitaille, R. Neuromuscular synaptogenesis: Coordinating partners with multiple functions. Nat. Rev. Neurosci. 2014, 15, 703–718. [Google Scholar] [CrossRef]
- Lopez-Font, I.; Sogorb-Esteve, A.; Javier-Torrent, M.; Brinkmalm, G.; Herrando-Grabulosa, M.; García-Lareu, B.; Turon-Sans, J.; Rojas-García, R.; Lleó, A.; Saura, C.A.; et al. Decreased circulating ErbB4 ectodomain fragments as a read-out of impaired signaling function in amyotrophic lateral sclerosis. Neurobiol. Dis. 2019, 124, 428–438. [Google Scholar] [CrossRef]
- Mancuso, R.; Martínez-Muriana, A.; Leiva, T.; Gregorio, D.; Ariza, L.; Morell, M.; Esteban-Pérez, J.; García-Redondo, A.; Calvo, A.C.; Atencia-Cibreiro, G.; et al. Neuregulin-1 promotes functional improvement by enhancing collateral sprouting in SOD1G93A ALS mice and after partial muscle denervation. Neurobiol. Dis. 2016, 95, 168–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Körner, S.; Thau-Habermann, N.; Kefalakes, E.; Bursch, F.; Petri, S. Expression of the axon-guidance protein receptor Neuropilin 1 is increased in the spinal cord and decreased in muscle of a mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2019, 49, 1529–1543. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wei-LaPierre, L.; Klose, A.; Dirksen, R.T.; Chakkalakal, J.V. Inducible depletion of adult skeletal muscle stem cells impairs the regeneration of neuromuscular junctions. Elife 2015, 4, e09221. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Klose, A.; Forman, S.; Paris, N.D.; Wei-LaPierre, L.; Cortés-Lopéz, M.; Tan, A.; Flaherty, M.; Miura, P.; Dirksen, R.T.; et al. Loss of adult skeletal muscle stem cells drives age-related neuromuscular junction degeneration. Elife 2017, 6, e26464. [Google Scholar] [CrossRef]
- Maimon, R.; Ionescu, A.; Bonnie, A.; Sweetat, S.; Wald-Altman, S.; Inbar, S.; Gradus, T.; Trotti, D.; Weil, M.; Behar, O.; et al. Mir126-5p downregulation facilitates axon degeneration and nmj disruption via a non–cell-autonomous mechanism in ALS. J. Neurosci. 2018, 38, 5478–5494. [Google Scholar] [CrossRef] [Green Version]
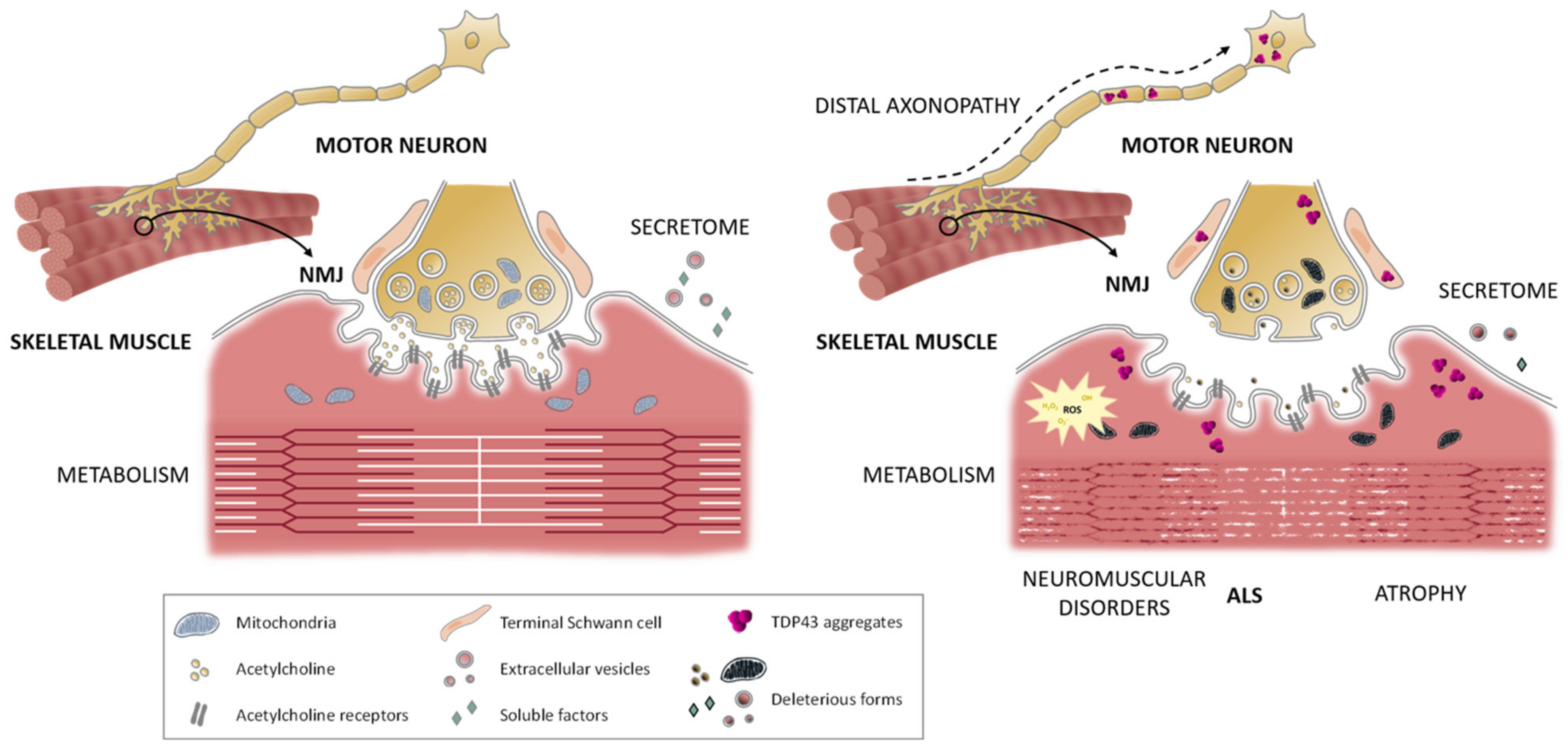

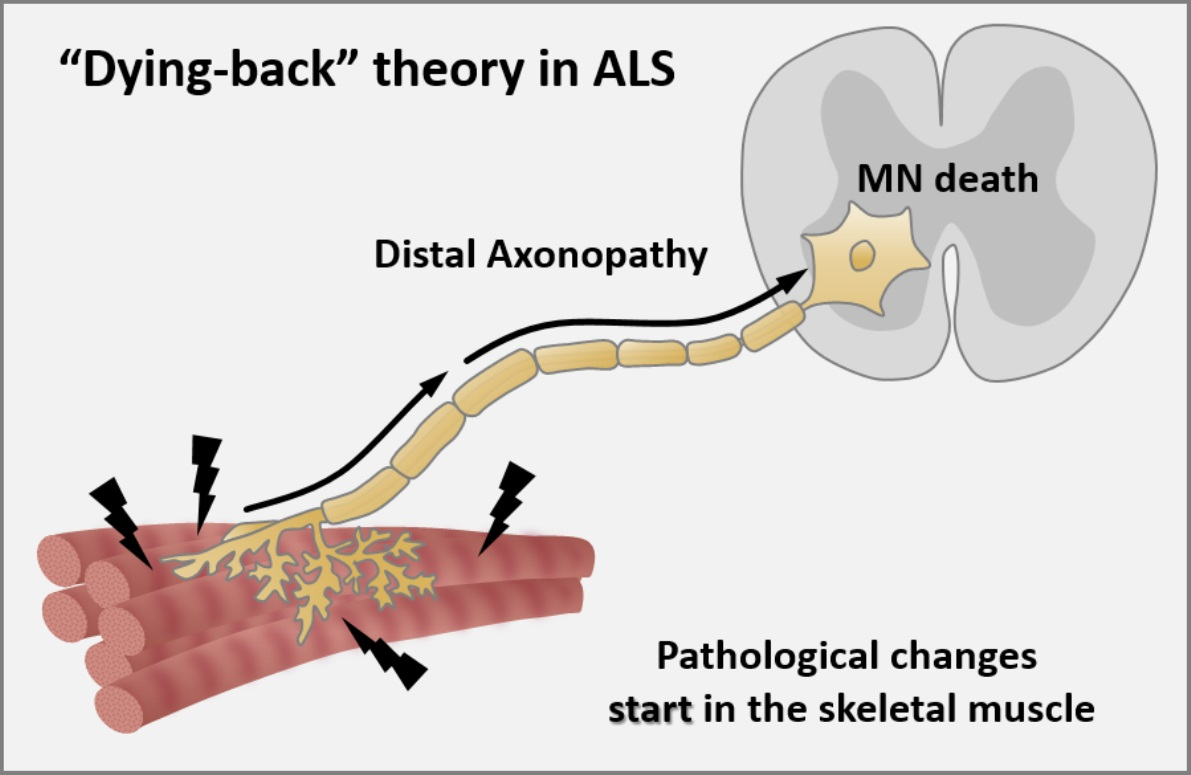{kind=link}
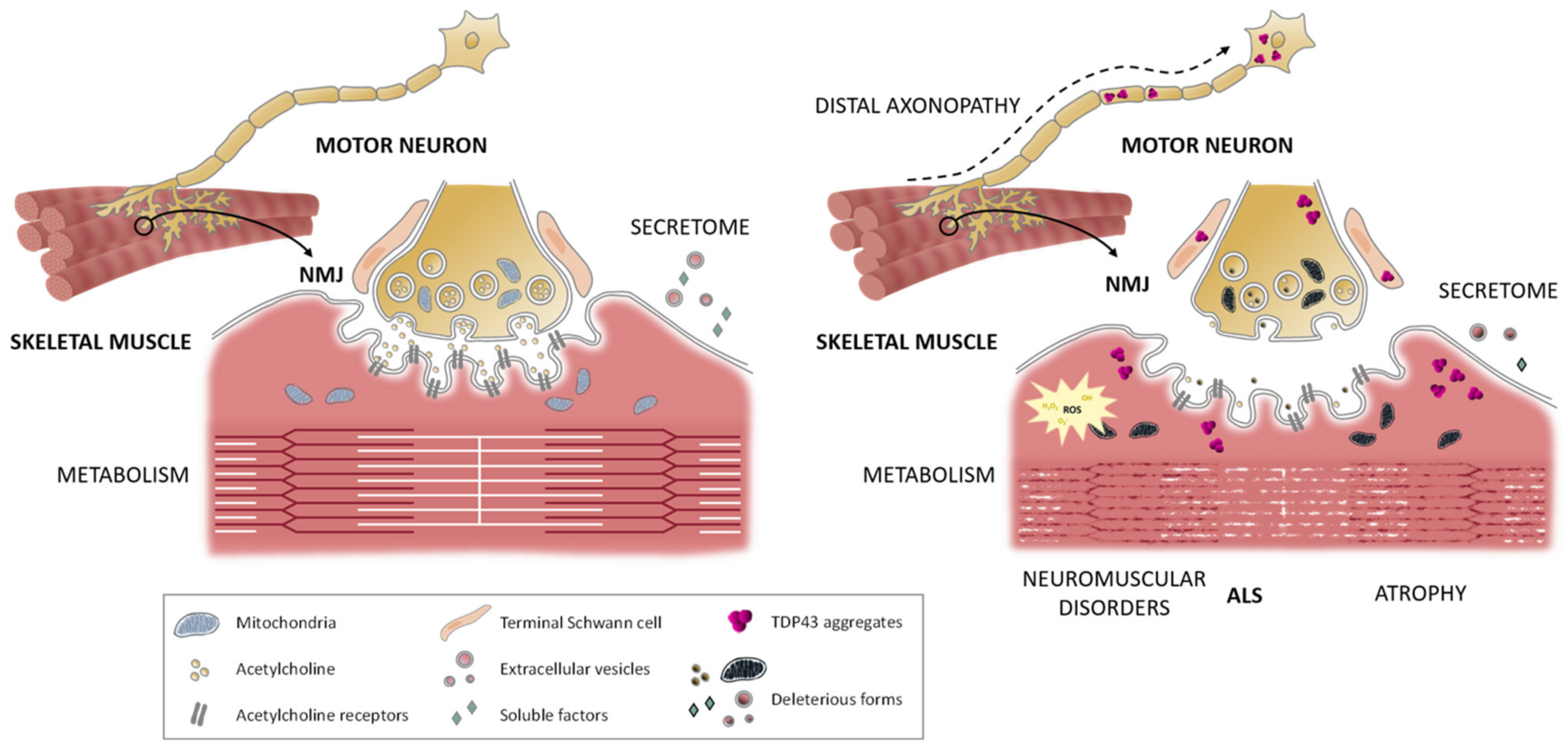{kind=link}
| Common Pathological Features | ||
|---|---|---|
| ALS | ||
| Structural | Motor neurons | Skeletal muscle |
| Decreased innervation | + (axonopathy) | + + + |
| NMJ dismantlement (fragmentation & loss) | + + + (pre-synaptic) | + + + (post-synaptic) |
| Reduced mass and strength / atrophy | + + + (spinal cord) | + + + |
| Cytoplasmic & nuclear aggregates | + + + | + + |
| ALS | ||
| Functional | Motor neurons | Skeletal muscle |
| Metabolic changes | + | + + + |
| Oxidative stress | + + + | + + |
| Deregulated nutrient sensing | + | + + |
| Loss of proteostasis | + + + | + + |
| Mitochondriopathy | + + | + |
| DNA damage | + + | + |
| Inflammation | Neuroinflammation | + |
| Altered intercellular communication | + + | + |
| Cellular senescence | + + | + (in vitro) |
| Cell death | + + + | + + |
| Muscle-Specific ALS Mouse Models | ||||||
|---|---|---|---|---|---|---|
| Mouse Lines | Tissue Specificity | ALS–Gene Regulation | Phenotypic/ Disease Onset | Muscle Phenotype | Neurological Phenotype | References |
| Acta1CRE;Sod1flox/flox (mSod1KO) | skeletal muscle (actin -1) | mouse Sod1 deletion | - Onset: 6–8 months - Survival: >16–17 months | No muscle atrophy Increase weakness Increased regenerative muscle fibers | No NMJ degeneration Not increased ROS production Not decreased mitochondrial ATP production | [88] |
| SOD1G93A/mIgf-1 (muscle rescue) | SOD1G93A ubiquitous Igf-1 in muscle | human SOD1-G93A overexpression and mouse Igf-1 overexpression | - G93A:onset: 90 days survival: <145 days - G93A/mIgf-1: onset: 110 days survival: <175 days | Maintenance of muscle integrity | NMJs stabilization Reduced inflammation in the spinal cord Enhanced MN survival | [89] |
| MLC/SOD1G93A | skeletal muscle (specific regulatory elements from MLC) | human SOD1-G93A overexpression | - Muscle atrophy: 4 weeks - Functional Performance: 16 weeks - Survival: not indicated | Muscle atrophy Significant reduction in muscle strength Alterations in the contractile apparatus Mitochondrial dysfunction Alteration in fiber type composition and metabolism (fast-to-slow shift) | NMJ dismantlement Microgliosis Hypomyelination in the sciatic nerve No MN loss in ventral spinal cord | [17,90,91,92] |
| WT-hSOD1mus G93A-hSOD1mus G37R-hSOD1mus | skeletal muscle (chicken -actin) | human SOD1 overexpression | Similar phenotype in all cases - Onset: 8–10 months - Survival: shortened 10–16% (slow disease progression) | Muscle atrophy Limb weakness and paresis Motor deficits Lifespan shortening Adipose tissue waste | Causes fatal ALS-like syndrome: Severe loss of NMJ Decreased innervation Axonopathy Spinal cord atrophy Loss of MN | [18,87] |
| TDP-43 TG | skeletal muscle (creatine kinase 8) | human TDP-43 overexpression | - Onset: 36 weeks - Survival: >18 months | Increased serum levels of myogenic enzymes Degenerative myofibers via ER stress Myotoxicity featuring tubular aggregates and TDP-43-positive inclusions | Not described | [93] |
| Pax7IREScreTardbpflox/WT | Pax7 lineage | 1 copy of mouse Tdp-43 deletion in MPCs and progeny | No phenotype in the adult but smaller myofibers after muscle damage-induced regeneration | TDP-43 is essential for skeletal-muscle-cell differentiation in culture and required for skeletal-muscle regeneration | Not described | [94] |
| MRL/MpJ | “super-healing” mouse model with siRNA | mouse Sod1 or Cat depletion (in vitro silencing of MPCs) | Great regenerative capacity for repair of many tissues | Impaired myogenic potential of MPCs A role for antioxidants in muscle repair | Not described | [80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pikatza-Menoio, O.; Elicegui, A.; Bengoetxea, X.; Naldaiz-Gastesi, N.; López de Munain, A.; Gerenu, G.; Gil-Bea, F.J.; Alonso-Martín, S. The Skeletal Muscle Emerges as a New Disease Target in Amyotrophic Lateral Sclerosis. J. Pers. Med. 2021, 11, 671. https://doi.org/10.3390/jpm11070671
Pikatza-Menoio O, Elicegui A, Bengoetxea X, Naldaiz-Gastesi N, López de Munain A, Gerenu G, Gil-Bea FJ, Alonso-Martín S. The Skeletal Muscle Emerges as a New Disease Target in Amyotrophic Lateral Sclerosis. Journal of Personalized Medicine. 2021; 11(7):671. https://doi.org/10.3390/jpm11070671
Chicago/Turabian StylePikatza-Menoio, Oihane, Amaia Elicegui, Xabier Bengoetxea, Neia Naldaiz-Gastesi, Adolfo López de Munain, Gorka Gerenu, Francisco Javier Gil-Bea, and Sonia Alonso-Martín. 2021. "The Skeletal Muscle Emerges as a New Disease Target in Amyotrophic Lateral Sclerosis" Journal of Personalized Medicine 11, no. 7: 671. https://doi.org/10.3390/jpm11070671
APA StylePikatza-Menoio, O., Elicegui, A., Bengoetxea, X., Naldaiz-Gastesi, N., López de Munain, A., Gerenu, G., Gil-Bea, F. J., & Alonso-Martín, S. (2021). The Skeletal Muscle Emerges as a New Disease Target in Amyotrophic Lateral Sclerosis. Journal of Personalized Medicine, 11(7), 671. https://doi.org/10.3390/jpm11070671