
Oxygen as a Master Regulator of Human Pluripotent Stem Cell Function and Metabolism



Abstract
:
1. Introduction
2. Basic Features of PSCs
3. Hypoxic Niche of PSCs
3.1. Oxygen Concentration Impacts the PSC Niche
3.2. Low Oxygen Condition during Embryonic Development
3.3. Hypoxia Affects hPSCs Culture In Vitro
4. Influence of Oxygen on iPSCs Metabolism
4.1. Glycolysis Promotes Pluripotency and Self-Renewal in PSCs
4.2. HIFs Mediate the Transcriptional Response to Low Oxygen Tension via Upregulation of Glycolysis
5. Oxygen on the Road to Pluripotency
5.1. Influence of Hypoxia on Reprogramming Efficiency
5.2. Metabolic Switch during Reprogramming
5.3. The Interplay between Core Transcription Factors at Different Stages of Reprograming
5.4. Role of HIFs in the Metabolic Switch during Reprogramming
5.5. Remodeling of Mitochondria
6. Metabolic Regulation of Pluripotency and Differentiation
6.1. Mitochondrial Dynamics Regulates Pluripotency and Differentiation in PSCs
6.2. OxPhos and ROS Production Are Emerging Mediators of Stem Cell Shift from Quiescence to Differentiation
6.3. Mitochondrial Regulatory Proteins Influence Cell Differentiation and Maturation of iPSCs
6.4. Bioenergetic Status of iPSCs Regulates Pluripotency and Differentiation
6.5. Nitric Oxide Drives Differentiation of PSCs via Mitochondrial Function
6.6. Acetyl-CoA Plays a Role in the Differentiation of PSCs via Chromatin Epigenetic Regulation
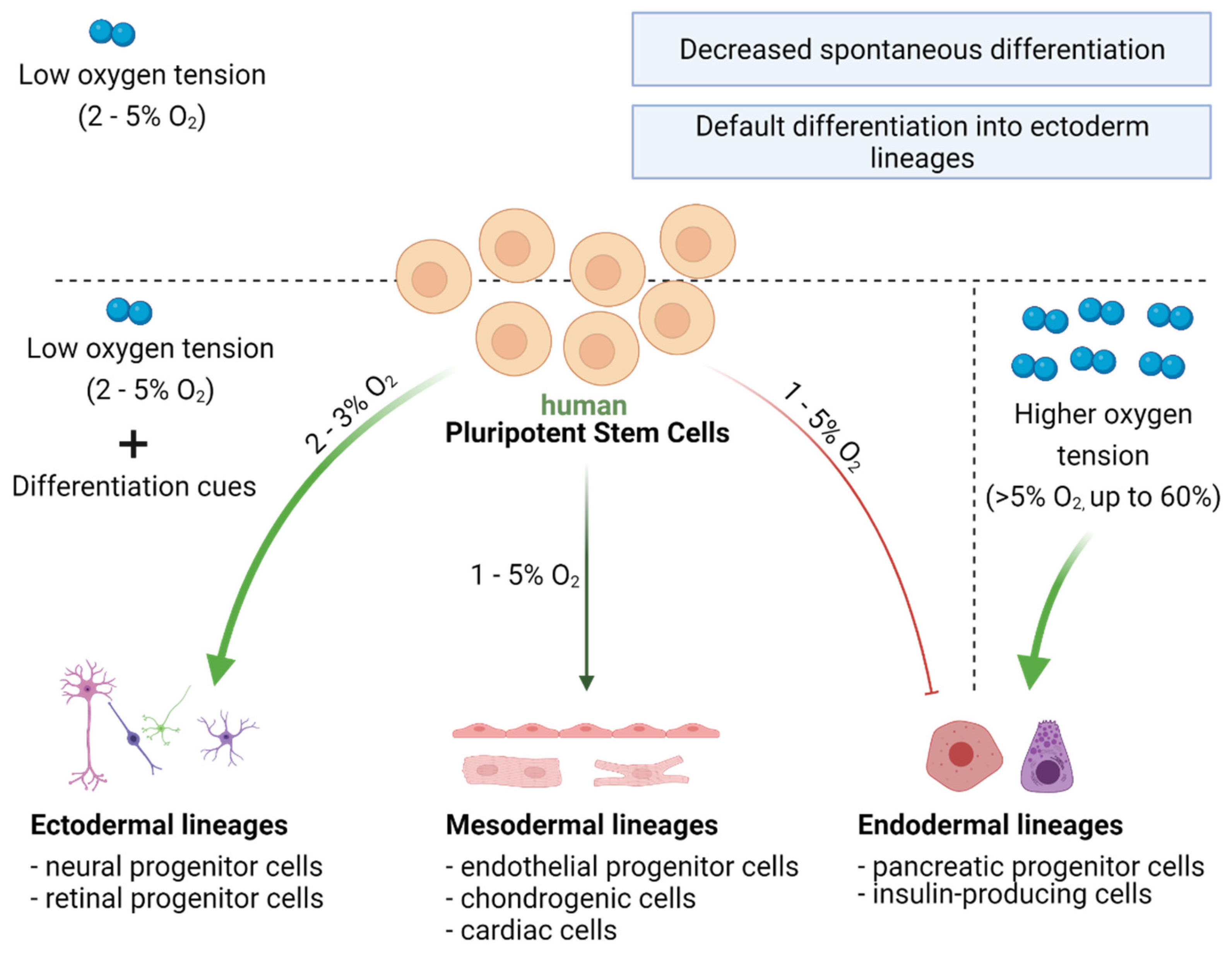
7. Oxygen Availability and Differentiation Pathways in PSCs
7.1. Oxygen Acts as a Morphogen during Development
7.2. Directed Differentiation of hPSCs Can Be Modulated by Oxygen Concentration
7.2.1. Ectoderm Differentiation
7.2.2. Mesoderm Differentiation
7.2.3. Endoderm Differentiation
8. Oxygen in Cancer Stem Cells
8.1. PSCs and CSCs—Are They Distinct or Similar?
8.2. Hypoxia Triggers Aggressive Phenotypes of CSCs
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kolios, G.; Moodley, Y. Introduction to stem cells and regenerative medicine. Respiration 2012, 85, 3–10. [Google Scholar] [CrossRef]
- Young, R.A. Control of the embryonic stem cell state. Cell 2011, 144, 940–954. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, N.; Rao, M.S. A review of the methods for human iPSC derivation. Methods Mol. Biol. 2013, 997, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.S.; Shearer, R.L.; Capowski, E.E.; Wright, L.S.; Wallace, K.A.; McMillan, E.L.; Zhang, S.C.; Gamm, D.M. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 16698–16703. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.F.; Inoue-Yokoo, T.; Tan, K.S.; Lai, M.I.; Sugiyama, D. Hematopoietic cell differentiation from embryonic and induced pluripotent stem cells. Stem Cell Res. Ther. 2013, 4, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chlebanowska, P.; Tejchman, A.; Sułkowski, M.; Skrzypek, K.; Majka, M. Use of 3D organoids as a model to study idiopathic form of parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Crespo, M.; Vilar, E.; Tsai, S.Y.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Witherspoon, M.; et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 2017, 23, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous induced stem-cell–derived retinal cells for macular degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Jehuda, R.; Shemer, Y.; Binah, O. Genome editing in induced pluripotent stem cells using CRISPR/Cas9. Stem Cell Rev. Rep. 2018, 14, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Li, Z.; Fung, H.-L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Lee, A.S.; Volkmer, J.P.; Sahoo, D.; Nag, D.; Mosley, A.R.; Inlay, M.A.; Ardehali, R.; Chavez, S.L.; Pera, R.R.; et al. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat. Biotechnol. 2011, 29, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Martins-Taylor, K.; Xu, R.-H. Concise review: Genomic stability of human induced pluripotent stem cells. Stem Cells 2012, 30, 22–27. [Google Scholar] [CrossRef]
- Närvä, E.; Pursiheimo, J.-P.; Laiho, A.; Rahkonen, N.; Emani, M.R.; Viitala, M.; Laurila, K.; Sahla, R.; Lund, R.; Lähdesmäki, H.; et al. Continuous hypoxic culturing of human embryonic stem cells enhances SSEA-3 and MYC levels. PLoS ONE 2013, 8, e78847. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, K.; Matsuura, T.; Nakazono, A.; Igawa, K.; Yamada, S.; Hayashi, Y. Effects of hypoxia inducible factors on pluripotency in human iPS cells. Microsc. Res. Tech. 2018, 81, 749–754. [Google Scholar] [CrossRef]
- Prigione, A.; Rohwer, N.; Hoffmann, S.; Mlody, B.; Drews, K.; Bukowiecki, R.; Blümlein, K.; Wanker, E.E.; Ralser, M.; Cramer, T.; et al. HIF1α modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells 2014, 32, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Iida, K.; Takeda-Kawaguchi, T.; Hada, M.; Yuriguchi, M.; Aoki, H.; Tamaoki, N.; Hatakeyama, D.; Kunisada, T.; Shibata, T.; Tezuka, K. Hypoxia-enhanced derivation of iPSCs from human dental pulp cells. J. Dent. Res. 2013, 92, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Kumar, N.; Kalsan, M.; Saini, A.; Chandra, R. Mechanism of induction: Induced pluripotent stem cells (iPSCs). J. Stem Cells 2015, 10, 43–62. [Google Scholar]
- Zhou, Q.; Hong, Y.; Zhan, Q.; Shen, Y.; Liu, Z. Role for Kruppel-like factor 4 in determining the outcome of p53 response to DNA damage. Cancer Res. 2009, 69, 8284–8292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gafni, O.; Weinberger, L.; Mansour, A.A.; Manor, Y.S.; Chomsky, E.; Ben-Yosef, D.; Kalma, Y.; Viukov, S.; Maza, I.; Zviran, A.; et al. Derivation of novel human ground state naive pluripotent stem cells. Nature 2013, 504, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.; Hockenbery, D.; Ware, C.; et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 2010, 28, 721–733. [Google Scholar] [CrossRef]
- Zhu, S.; Li, W.; Zhou, H.; Wei, W.; Ambasudhan, R.; Lin, T.; Kim, J.; Zhang, K.; Ding, S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 2010, 7, 651–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishwakarma, A.; Rouwkema, J.; Jones, P.A.; Karp, J.M. The Need to Study, Mimic, and Target Stem Cell Niches; Elsevier Inc.: Amsterdam, The Netherlands, 2017; ISBN 9780128027561. [Google Scholar]
- Mohyeldin, A.; Garzón-Muvdi, T.; Quiñones-Hinojosa, A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosová, I.; Dao, M.; Capoccia, B.; Link, D.; Nolta, J.A. Hypoxic preconditioning results in increased motility and improved therapeutic potential of human mesenchymal stem cells. Stem Cells 2008, 26, 2173–2182. [Google Scholar] [CrossRef] [Green Version]
- Eliasson, P.; Jönsson, J.-I. The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J. Cell. Physiol. 2010, 222, 17–22. [Google Scholar] [CrossRef]
- Kubota, Y.; Takubo, K.; Suda, T. Bone marrow long label-retaining cells reside in the sinusoidal hypoxic niche. Biochem. Biophys. Res. Commun. 2008, 366, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Santilli, G.; Lamorte, G.; Carlessi, L.; Ferrari, D.; Nodari, L.R.; Binda, E.; Delia, D.; Vescovi, A.L.; De Filippis, L. Mild hypoxia enhances proliferation and multipotency of human neural stem cells. PLoS ONE 2010, 5, e8575. [Google Scholar] [CrossRef]
- Yoshida, Y.; Takahashi, K.; Okita, K.; Ichisaka, T.; Yamanaka, S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 2009, 5, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezashi, T.; Das, P.; Roberts, R.M. Low O2 tensions and the prevention of differentiation of hES cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4783–4788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsyth, N.R.; Musio, A.; Vezzoni, P.; Simpson, A.H.R.W.; Noble, B.S.; McWhir, J. Physiologic oxygen enhances human embryonic stem cell clonal recovery and reduces chromosomal abnormalities. Cloning Stem Cells 2006, 8, 16–23. [Google Scholar] [CrossRef]
- Lim, H.-J.; Han, J.; Woo, D.-H.; Kim, S.-E.; Kim, S.K.; Kang, H.-G.; Kim, J.-H. Biochemical and morphological effects of hypoxic environment on human embryonic stem cells in long-term culture and differentiating embryoid bodies. Mol. Cells 2011, 31, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, M.; Bhatia, M. Pluripotent Stem Cell Microenvironment; Elsevier Inc.: Amsterdam, The Netherlands, 2017; ISBN 9780128027561. [Google Scholar]
- Forristal, C.E.; Wright, K.L.; Hanley, N.A.; Oreffo, R.O.C.; Houghton, F.D. Hypoxia inducible factors regulate pluripotency and proliferation in human embryonic stem cells cultured at reduced oxygen tensions. Reproduction 2010, 139, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Bavister, B. Oxygen concentration and preimplantation development. Reprod. Biomed. Online 2004, 9, 484–486. [Google Scholar] [CrossRef]
- Fischer, B.; Bavister, B.D. Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J. Reprod. Fertil. 1993, 99, 673–679. [Google Scholar] [CrossRef]
- Karagenc, L.; Sertkaya, Z.; Ciray, N.; Ulug, U.; Bahçeci, M. Impact of oxygen concentration on embryonic development of mouse zygotes. Reprod. Biomed. Online 2004, 9, 409–417. [Google Scholar] [CrossRef]
- Braun, R.D.; Lanzen, J.L.; Snyder, S.A.; Dewhirst, M.W. Comparison of tumor and normal tissue oxygen tension measurements using OxyLite or microelectrodes in rodents. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2533–H2544. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.J.; Kind, K.L.; Pantaleon, M.; Armstrong, D.T.; Thompson, J.G. Oxygen-regulated gene expression in bovine blastocysts. Biol. Reprod. 2004, 71, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-F.; Kuo, H.-C.; Chen, W.; Wu, F.-C.; Yang, Y.-S.; Ho, H.-N. A reduced oxygen tension (5%) is not beneficial for maintaining human embryonic stem cells in the undifferentiated state with short splitting intervals. Hum. Reprod. 2009, 24, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengner, C.J.; Gimelbrant, A.A.; Erwin, J.A.; Cheng, A.W.; Guenther, M.G.; Welstead, G.G.; Alagappan, R.; Frampton, G.M.; Xu, P.; Muffat, J.; et al. Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell 2010, 141, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.-W.; Kawakatsu, M.; Idemitsu, M.; Urata, Y.; Goto, S.; Ono, Y.; Hamano, K.; Li, T.S. Culture under low physiological oxygen conditions improves the stemness and quality of induced pluripotent stem cells. J. Cell. Physiol. 2013, 228, 2159–2166. [Google Scholar] [CrossRef]
- Westfall, S.D.; Sachdev, S.; Das, P.; Hearne, L.B.; Hannink, M.; Roberts, R.M.; Ezashi, T. Identification of oxygen-sensitive transcriptional programs in human embryonic stem cells. Stem Cells Dev. 2008, 17, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, N.R.; Kay, A.; Hampson, K.; Downing, A.; Talbot, R.; McWhir, J. Transcriptome alterations due to physiological normoxic (2% O2) culture of human embryonic stem cells. Regen. Med. 2008, 3, 817–833. [Google Scholar] [CrossRef]
- Gu, W.; Gaeta, X.; Sahakyan, A.; Chan, A.B.; Hong, C.S.; Kim, R.; Braas, D.; Plath, K.; Lowry, W.E.; Christofk, H.R. Glycolytic metabolism plays a functional role in regulating human pluripotent stem cell state. Cell Stem Cell 2016, 19, 476–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Jang, H.; Kim, T.W.; Kang, B.H.; Lee, S.E.; Jeon, Y.K.; Chung, D.H.; Choi, J.; Shin, J.; Cho, E.-J.; et al. Core pluripotency factors directly regulate metabolism in embryonic stem cell to maintain pluripotency. Stem Cells 2015, 33, 2699–2711. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.G.; Cliff, T.S.; Gammilonghi, A.; Ryall, J.G.; Dalton, S.; Gardner, D.K.; Harvey, A.J. Oxygen regulates human pluripotent stem cell metabolic flux. Stem Cells Int. 2019, 2019, 8195614. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Izpisua Belmonte, J.C. Anaerobicizing into pluripotency. Cell Metab. 2011, 14, 143–144. [Google Scholar] [CrossRef] [Green Version]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting metabolism for cancer therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohyama, S.; Fujita, J.; Hishiki, T.; Matsuura, T.; Hattori, F.; Ohno, R.; Kanazawa, H.; Seki, T.; Nakajima, K.; Kishino, Y.; et al. Glutamine oxidation is indispensable for survival of human pluripotent stem cells. Cell Metab. 2016, 23, 663–674. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. A Time for MYC: Metabolism and therapy. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Mathieu, J.; Zhou, W.; Xing, Y.; Sperber, H.; Ferreccio, A.; Agoston, Z.; Kuppusamy, K.T.; Moon, R.T.; Ruohola-Baker, H. Hypoxia-inducible factors have distinct and stage-specific roles during reprogramming of human cells to pluripotency. Cell Stem Cell 2014, 14, 592–605. [Google Scholar] [CrossRef] [Green Version]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Varlakhanova, N.V.; Cotterman, R.F.; deVries, W.N.; Morgan, J.; Donahue, L.R.; Murray, S.; Knowles, B.B.; Knoepfler, P.S. Myc maintains embryonic stem cell pluripotency and self-renewal. Differentiation 2010, 80, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Kenneth, N.S.; Rocha, S. Regulation of gene expression by hypoxia. Biochem. J. 2008, 414, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-X.; Lin, P.-H.; Rahmawati, E.; Ma, Y.-Y.; Chan, C.; Tzeng, C.-R. Mitochondria Research in Human Reproduction in the Ovary, 3rd ed.Academic Press: Cambridge, MA, USA, 2019; pp. 327–335. ISBN 9780128132098. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, J.; Rafiee, M.-R.; Reiland, S.; Polo, J.M.; Gehring, J.; Okawa, S.; Huber, W.; Hochedlinger, K.; Krijgsveld, J. Highly coordinated proteome dynamics during reprogramming of somatic cells to pluripotency. Cell Rep. 2012, 2, 1579–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerías, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012, 22, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Cacchiarelli, D.; Trapnell, C.; Ziller, M.J.; Soumillon, M.; Cesana, M.; Karnik, R.; Donaghey, J.; Smith, Z.D.; Ratanasirintrawoot, S.; Zhang, X.; et al. Integrative analyses of human reprogramming reveal dynamic nature of induced pluripotency. Cell 2015, 162, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Folmes, C.D.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Prieto, J.; Seo, A.Y.; León, M.; Santacatterina, F.; Torresano, L.; Palomino-Schätzlein, M.; Giménez, K.; Vallet-Sánchez, A.; Ponsoda, X.; Pineda-Lucena, A.; et al. MYC induces a hybrid energetics program early in cell reprogramming. Stem Cell Rep. 2018, 11, 1479–1492. [Google Scholar] [CrossRef] [Green Version]
- Kida, Y.S.; Kawamura, T.; Wei, Z.; Sogo, T.; Jacinto, S.; Shigeno, A.; Kushige, H.; Yoshihara, E.; Liddle, C.; Ecker, J.R.; et al. ERRs mediate a metabolic switch required for somatic cell reprogramming to pluripotency. Cell Stem Cell 2015, 16, 547–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, J.; León, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; López-Garciá, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef]
- Ji, J.; Sharma, V.; Qi, S.; Guarch, M.E.; Zhao, P.; Luo, Z.; Fan, W.; Wang, Y.; Mbabaali, F.; Neculai, D.; et al. Antioxidant supplementation reduces genomic aberrations in human induced pluripotent stem cells. Stem Cell Rep. 2014, 2, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, K.E.; Joy, S.; Delhove, J.M.K.M.; Kotiadis, V.N.; Fernandez, E.; Fitzpatrick, L.M.; Whiteford, J.R.; King, P.J.; Bolanos, J.P.; Duchen, M.R.; et al. NRF2 orchestrates the metabolic shift during induced pluripotent stem cell reprogramming. Cell Rep. 2016, 14, 1883–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, K.; Aizawa, S.; Nugroho, F.L.; Shiomitsu, E.; Tran, Y.T.H.; Bui, P.L.; Borisova, E.; Sakuragi, Y.; Takada, H.; Kurisaki, A.; et al. A Role for KLF4 in promoting the metabolic shift via TCL1 during induced pluripotent stem cell generation. Stem Cell Rep. 2017, 8, 787–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, B.D.; Bernards, R.; Peeper, D.S. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat. Cell Biol. 2005, 7, 1074–1082. [Google Scholar] [CrossRef]
- Topham, C.; Tighe, A.; Ly, P.; Bennett, A.; Sloss, O.; Nelson, L.; Ridgway, R.A.; Huels, D.; Littler, S.; Schandl, C.; et al. MYC is a major determinant of mitotic cell fate. Cancer Cell 2015, 28, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Nakao, S.; Ueyama, T.; Harada, Y.; Kawamura, T. Metabolic remodeling during somatic cell reprogramming to induced pluripotent stem cells: Involvement of hypoxia-inducible factor 1. Inflamm. Regen. 2020, 40, 8. [Google Scholar] [CrossRef]
- Xu, X.; Duan, S.; Yi, F.; Ocampo, A.; Liu, G.H.; Izpisua Belmonte, J.C. Mitochondrial regulation in pluripotent stem cells. Cell Metab. 2013, 18, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S. Biological roles of alternative autophagy. Mol. Cells 2018, 41, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xia, P.; Ye, B.; Huang, G.; Liu, J.; Fan, Z. Transient activation of autophagy via Sox2-mediated suppression of mTOR is an important early step in reprogramming to pluripotency. Cell Stem Cell 2013, 13, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Cui, P.; Cai, Y.; Wang, L.; He, X.; Long, P.; Lu, K.; Yan, R.; Zhang, Y.; Pan, X.; et al. Mitochondrial dynamics is critical for the full pluripotency and embryonic developmental potential of pluripotent stem cells. Cell Metab. 2019, 29, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Fan, L.; Cen, P.; Chen, E.; Jiang, Z.; Li, L. Energy metabolism plays a critical role in stem cell maintenance and differentiation. Int. J. Mol. Sci. 2016, 17, 253. [Google Scholar] [CrossRef] [Green Version]
- Mattenberger, Y.; James, D.I.; Martinou, J.-C. Fusion of mitochondria in mammalian cells is dependent on the mitochondrial inner membrane potential and independent of microtubules or actin. FEBS Lett. 2003, 538, 53–59. [Google Scholar] [CrossRef]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, S.; Lin, Y.-C.; Tsai, M.-H.; Lin, C.-S.; Murayama, Y.; Sato, R.; Yokoyama, K.K. Emerging roles of hypoxia-inducible factors and reactive oxygen species in cancer and pluripotent stem cells. Kaohsiung J. Med. Sci. 2015, 31, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Cornacchia, D.; Zhang, C.; Zimmer, B.; Chung, S.Y.; Fan, Y.; Soliman, M.A.; Tchieu, J.; Chambers, S.M.; Shah, H.; Paull, D.; et al. Lipid deprivation induces a stable, naive-to-primed intermediate state of pluripotency in human PSCs. Cell Stem Cell 2019, 25, 120–136. [Google Scholar] [CrossRef]
- Kanno, S.; Kim, P.K.; Sallam, K.; Lei, J.; Billiar, T.R.; Shears, L.L., 2nd. Nitric oxide facilitates cardiomyogenesis in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2004, 101, 12277–12281. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Bai, H.; Guo, F.; Thai, P.N.; Luo, X.; Zhang, P.; Yang, C.; Feng, X.; Zhu, D.; Guo, J.; et al. PGC-1α activator ZLN005 promotes maturation of cardiomyocytes derived from human embryonic stem cells. Aging 2020, 12, 7411–7430. [Google Scholar] [CrossRef]
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014, 32, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.-C.; Wu, Y.-T.; Tsai, C.-L.; Wei, Y.-H. Current understanding and future perspectives of the roles of sirtuins in the reprogramming and differentiation of pluripotent stem cells. Exp. Biol. Med. 2018, 243, 563–575. [Google Scholar] [CrossRef]
- Jing, R.; Corbett, J.L.; Cai, J.; Beeson, G.C.; Beeson, C.C.; Chan, S.S.; Dimmock, D.P.; Lazcares, L.; Geurts, A.M.; Lemasters, J.J.; et al. A screen using iPSC-derived hepatocytes reveals NAD + as a potential treatment for mtDNA depletion syndrome. Cell Rep. 2018, 25, 1469–1484.e5. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Martin, A.; Vellon, L.; Quirós, P.M.; Cufí, S.; Ruiz de Galarreta, E.; Oliveras-Ferraros, C.; Martin, A.G.; Martin-Castillo, B.; López-Otín, C.; Menendez, J.A. Activation of AMP-activated protein kinase (AMPK) provides a metabolic barrier to reprogramming somatic cells into stem cells. Cell Cycle 2012, 11, 974–989. [Google Scholar] [CrossRef] [Green Version]
- Tejedo, J.R.; Tapia-Limonchi, R.; Mora-Castilla, S.; Cahuana, G.M.; Hmadcha, A.; Martin, F.; Bedoya, F.J.; Soria, B. Low concentrations of nitric oxide delay the differentiation of embryonic stem cells and promote their survival. Cell Death Dis 2010, 1, e80. [Google Scholar] [CrossRef] [Green Version]
- Takashima, Y.; Guo, G.; Loos, R.; Nichols, J.; Ficz, G.; Krueger, F.; Oxley, D.; Santos, F.; Clarke, J.; Mansfield, W.; et al. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell 2014, 158, 1254–1269. [Google Scholar] [CrossRef] [Green Version]
- Shyh-Chang, N.; Ng, H.-H. The metabolic programming of stem cells. Genes Dev. 2017, 31, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular a-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, H.E.; Lo, J.; Johnson, R.S. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998, 17, 3005–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia- inducible factor 1α. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prado-Lopez, S.; Conesa, A.; Armiñán, A.; Martínez-Losa, M.; Escobedo-Lucea, C.; Gandia, C.; Tarazona, S.; Melguizo, D.; Blesa, D.; Montaner, D.; et al. Hypoxia promotes efficient differentiation of human embryonic stem cells to functional endothelium. Stem Cells 2010, 28, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, J.; Lin, Y.; Gaeta, X.; Meng, X.; Wisidagama, D.R.R.; Cinkornpumin, J.; Koehler, C.M.; Malone, C.S.; Teitell, M.A.; et al. Defining the role of oxygen tension in human neural progenitor fate. Stem Cell Rep. 2014, 3, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Fynes, K.; Tostoes, R.; Ruban, L.; Weil, B.; Mason, C.; Veraitch, F.S. The differential effects of 2% oxygen preconditioning on the subsequent differentiation of mouse and human pluripotent stem cells. Stem Cells Dev. 2014, 23, 1910–1922. [Google Scholar] [CrossRef]
- Cui, P.; Zhang, P.; Zhang, Y.; Sun, L.; Cui, G.; Guo, X.; Wang, H.; Zhang, X.; Shi, Y.; Yu, Z. HIF-1α/Actl6a/H3K9ac axis is critical for pluripotency and lineage differentiation of human induced pluripotent stem cells. FASEB J. 2020, 34, 5740–5753. [Google Scholar] [CrossRef]
- Risau, W.; Flamme, I. Vasculogenesis. Annu. Rev. Cell Dev. Biol. 1995, 11, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, S.R.L.; Bilican, B.; Webber, D.J.; Luzhynskaya, A.; He, X.L.; Compston, A.; Karadottir, R.; Franklin, R.J.M.; Chandran, S. Derivation of neural precursor cells from human ES cells at 3% O2 is efficient, enhances survival and presents no barrier to regional specification and functional differentiation. Cell Death Differ. 2011, 18, 1016–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, R.; Onodera, K.; Ito, T.; Doyu, M.; Okano, H.J.; Okada, Y. Modulation of oxygen tension, acidosis, and cell density is crucial for neural differentiation of human induced pluripotent stem cells. Neurosci. Res. 2021, 163, 34–42. [Google Scholar] [CrossRef]
- Bae, D.; Mondragon-Teran, P.; Hernandez, D.; Ruban, L.; Mason, C.; Bhattacharya, S.S.; Veraitch, F.S. Hypoxia enhances the generation of retinal progenitor cells from human induced pluripotent and embryonic stem cells. Stem Cells Dev. 2012, 21, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Niebruegge, S.; Bauwens, C.L.; Peerani, R.; Thavandiran, N.; Masse, S.; Sevaptisidis, E.; Nanthakumar, K.; Woodhouse, K.; Husain, M.; Kumacheva, E.; et al. Generation of human embryonic stem cell-derived mesoderm and cardiac cells using size-specified aggregates in an oxygen-controlled bioreactor. Biotechnol. Bioeng. 2009, 102, 493–507. [Google Scholar] [CrossRef]
- Koay, E.J.; Athanasiou, K.A. Hypoxic chondrogenic differentiation of human embryonic stem cells enhances cartilage protein synthesis and biomechanical functionality. Osteoarthr. Cartil. 2008, 16, 1450–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, P.W.; Thompson, S.; Millrod, M.A.; Weinberg, S.; Yuan, X.; Peters, A.; Mahairaki, V.; Koliatsos, V.E.; Tung, L.; Zambidis, E.T. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS ONE 2011, 6, e18293. [Google Scholar] [CrossRef] [PubMed]
- Podkalicka, P.; Stępniewski, J.; Mucha, O.; Kachamakova-Trojanowska, N.; Dulak, J.; Łoboda, A. Hypoxia as a driving force of pluripotent stem cell reprogramming and differentiation to endothelial cells. Biomolecules 2020, 10, 1614. [Google Scholar] [CrossRef]
- Bobis-Wozowicz, S.; Kmiotek-Wasylewska, K.; Madeja, Z.; Zuba-Surma, E. Hypoxia pre-conditioning enhances cardiac differentiation ability of human iPSCs. Hum. Gene Ther. 2017, 28, A92. [Google Scholar]
- Hakim, F.; Kaitsuka, T.; Raeed, J.M.; Wei, F.-Y.; Shiraki, N.; Akagi, T.; Yokota, T.; Kume, S.; Tomizawa, K. High oxygen condition facilitates the differentiation of mouse and human pluripotent stem cells into pancreatic progenitors and insulin-producing cells. J. Biol. Chem. 2014, 289, 9623–9638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Afzali, M.; Vatankhah, M.; Ostad, S.N. Investigation of simvastatin-induced apoptosis and cell cycle arrest in cancer stem cells of MCF-7. J. Cancer Res. Ther. 2016, 12, 725–730. [Google Scholar] [CrossRef]
- Golan, H.; Shukrun, R.; Caspi, R.; Vax, E.; Pode-Shakked, N.; Goldberg, S.; Pleniceanu, O.; Bar-Lev, D.D.; Mark-Danieli, M.; Pri-Chen, S.; et al. In vivo expansion of cancer stemness affords novel cancer stem cell targets: Malignant rhabdoid tumor as an example. Stem Cell Rep. 2018, 11, 795–810. [Google Scholar] [CrossRef] [Green Version]
- Bahmad, H.F.; Cheaito, K.; Chalhoub, R.M.; Hadadeh, O.; Monzer, A.; Ballout, F.; El-Hajj, A.; Mukherji, D.; Liu, Y.N.; Daoud, G.; et al. Sphere-Formation assay: Three-dimensional in vitro culturing of prostate cancer stem/Progenitor sphere-forming cells. Front. Oncol. 2018, 8, 347. [Google Scholar] [CrossRef] [Green Version]
- Dianat-Moghadam, H.; Heydarifard, M.; Jahanban-Esfahlan, R.; Panahi, Y.; Hamishehkar, H.; Pouremamali, F.; Rahbarghazi, R.; Nouri, M. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J. Control. Release 2018, 288, 62–83. [Google Scholar] [CrossRef]
- Cao, S.; Wang, Z.; Gao, X.; He, W.; Cai, Y.; Chen, H.; Xu, R. FOXC1 induces cancer stem cell-like properties through upregulation of beta-catenin in NSCLC. J. Exp. Clin. Cancer Res. 2018, 37, 220. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.-Y.; Suzuki, H.; Honda, M.; Okada, H.; Kaneko, S.; Inoue, I.; Ebisui, E.; Hashimoto, K.; Carninci, P.; Kanki, K.; et al. Prevention of hepatocellular carcinoma by targeting MYCN-positive liver cancer stem cells with acyclic retinoid. Proc. Natl. Acad. Sci. USA 2018, 115, 4969–4974. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, O.; Knobloch, L.; Fornsaglio, J.; Varum, S.; Easley, C.; Schatten, G. DNA damage responses in human induced pluripotent stem cells and embryonic stem cells. PLoS ONE 2010, 5, e13410. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Robert, C.; Jang, Y.-Y.; Liu, H.; Sharkis, S.; Baylin, S.B.; Rassool, F.V. Human induced pluripotent cells resemble embryonic stem cells demonstrating enhanced levels of DNA repair and efficacy of nonhomologous end-joining. Mutat. Res. Mol. Mech. Mutagen. 2011, 713, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, H.; Weinstock, D.M. Repair at single targeted DNA double-strand breaks in pluripotent and differentiated human cells. PLoS ONE 2011, 6, e20514. [Google Scholar] [CrossRef] [PubMed]
- Mujoo, K.; Pandita, R.K.; Tiwari, A.; Charaka, V.; Chakraborty, S.; Singh, D.K.; Hambarde, S.; Hittelman, W.N.; Horikoshi, N.; Hunt, C.R.; et al. Differentiation of human induced pluripotent or embryonic stem cells decreases the DNA damage repair by homologous recombination. Stem Cell Rep. 2017, 9, 1660–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, F.; Wang, J.-H.; Fan, W.-J.; Meng, Y.-T.; Li, M.-M.; Li, T.-T.; Cui, B.; Wang, H.-F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; Della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’avella, D.; Basso, G. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells 2010, 28, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.-W.; Tong, G.-H.; Liu, Y. Cancer stem cells and hypoxia-inducible factors. Int. J. Oncol. 2018, 53, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef] [Green Version]
- Bertout, J.A.; Majmundar, A.J.; Gordan, J.D.; Lam, J.C.; Ditsworth, D.; Keith, B.; Brown, E.J.; Nathanson, K.L.; Simon, M.C. HIF2α inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc. Natl. Acad. Sci. USA 2009, 106, 14391–14396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordan, J.D.; Bertout, J.A.; Hu, C.-J.; Diehl, J.A.; Simon, M.C. HIF-2α Promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, N.; Koritzinsky, M.; Zhao, H.; Bindra, R.; Glazer, P.M.; Powell, S.; Belmaaza, A.; Wouters, B.; Bristow, R.G. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res 2008, 68, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Olcina, M.; Lecane, P.S.; Hammond, E.M. Targeting hypoxic cells through the DNA damage response. Clin. Cancer Res. 2010, 16, 5624–5629. [Google Scholar] [CrossRef] [Green Version]
- Krishnamachary, B.; Berg-Dixon, S.; Kelly, B.; Agani, F.; Feldser, D.; Ferreira, G.; Iyer, N.; LaRusch, J.; Pak, B.; Taghavi, P.; et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003, 63, 1138–1143. [Google Scholar]
- Choi, Y.-J.; Shin, H.-W.; Chun, Y.-S.; Leutou, A.S.; Son, B.W.; Park, J.-W. Diacetoxyscirpenol as a new anticancer agent to target hypoxia-inducible factor 1. Oncotarget 2016, 7, 62107–62122. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.-P.; Wong, C.C.L.; Kai, A.K.-L.; Ho, D.W.H.; Lau, E.Y.T.; Tsui, Y.-M.; Chan, L.-K.; Cheung, T.-T.; Chok, K.S.H.; Chan, A.C.Y.; et al. SENP1 promotes hypoxia-induced cancer stemness by HIF-1α deSUMOylation and SENP1/HIF-1α positive feedback loop. Gut 2017, 66, 2149–2159. [Google Scholar] [CrossRef]
- Jiang, N.; Zou, C.; Zhu, Y.; Luo, Y.; Chen, L.; Lei, Y.; Tang, K.; Sun, Y.; Zhang, W.; Li, S.; et al. HIF-1ɑ-regulated miR-1275 maintains stem cell-like phenotypes and promotes the progression of LUAD by simultaneously activating Wnt/β-catenin and Notch signaling. Theranostics 2020, 10, 2553–2570. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | No. of Cell Lines | % O2 | Duration of H | Substrate | Sponta-Neous Differen-Tiation | Cell Growth | Markers of Pluripo-Tency | Additional Remarks | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| hESCs | 1 | 1; 3; 5 | 15 d | MEF; Ma | ↓ | ↓ | nd | enhanced EBs formation | [36] |
| hESCs | 1 | 5 | 3 p | MEF; Ma | ↓ | ↑ | ↑ | HIF2α as the most important regulator in H | [40] |
| hESCs | 3 | 4 | 7 d | hfF | ↓ | ↑ | ↑/↔ | increased MYC expression via HIF2α | [19] |
| hESCs | 3 | 2 | 10 p | Ma | nd | nd | ↔ | 302 genes up in H (transcriptome analysis) | [50] |
| hESCs | 3 | 2 | 5 p | Ma | nd | ↑ | ↔ | smaller and less granular cells; reduced chromosomal aberrations | [37] |
| hESCs | 2 | 5 | 7; 14; 28 d | MEF | ↓ | ↓ | ↔ | - | [46] |
| hESCs | 2 | 4 | 10 p | MEF; Ma | ↔ | nd | ↔ | - | [49] |
| hiPSCs | 2 | 2.5; 5 | 2 m | MEF | nd | nd | ↑ NANOG ↓ OCT4 | 53PB1 down | [48] |
| hiPSCs | 1 | 5 | 14 d | MEF | ↓ | ↑ | ↑ | HIF2α activated in H | [20] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nit, K.; Tyszka-Czochara, M.; Bobis-Wozowicz, S. Oxygen as a Master Regulator of Human Pluripotent Stem Cell Function and Metabolism. J. Pers. Med. 2021, 11, 905. https://doi.org/10.3390/jpm11090905
Nit K, Tyszka-Czochara M, Bobis-Wozowicz S. Oxygen as a Master Regulator of Human Pluripotent Stem Cell Function and Metabolism. Journal of Personalized Medicine. 2021; 11(9):905. https://doi.org/10.3390/jpm11090905
Chicago/Turabian StyleNit, Kinga, Malgorzata Tyszka-Czochara, and Sylwia Bobis-Wozowicz. 2021. "Oxygen as a Master Regulator of Human Pluripotent Stem Cell Function and Metabolism" Journal of Personalized Medicine 11, no. 9: 905. https://doi.org/10.3390/jpm11090905
APA StyleNit, K., Tyszka-Czochara, M., & Bobis-Wozowicz, S. (2021). Oxygen as a Master Regulator of Human Pluripotent Stem Cell Function and Metabolism. Journal of Personalized Medicine, 11(9), 905. https://doi.org/10.3390/jpm11090905