
Integrating Genetic Alterations and the Hippo Pathway in Head and Neck Squamous Cell Carcinoma for Future Precision Medicine
 , , , ,
, , , , 
Abstract
:1. Introduction
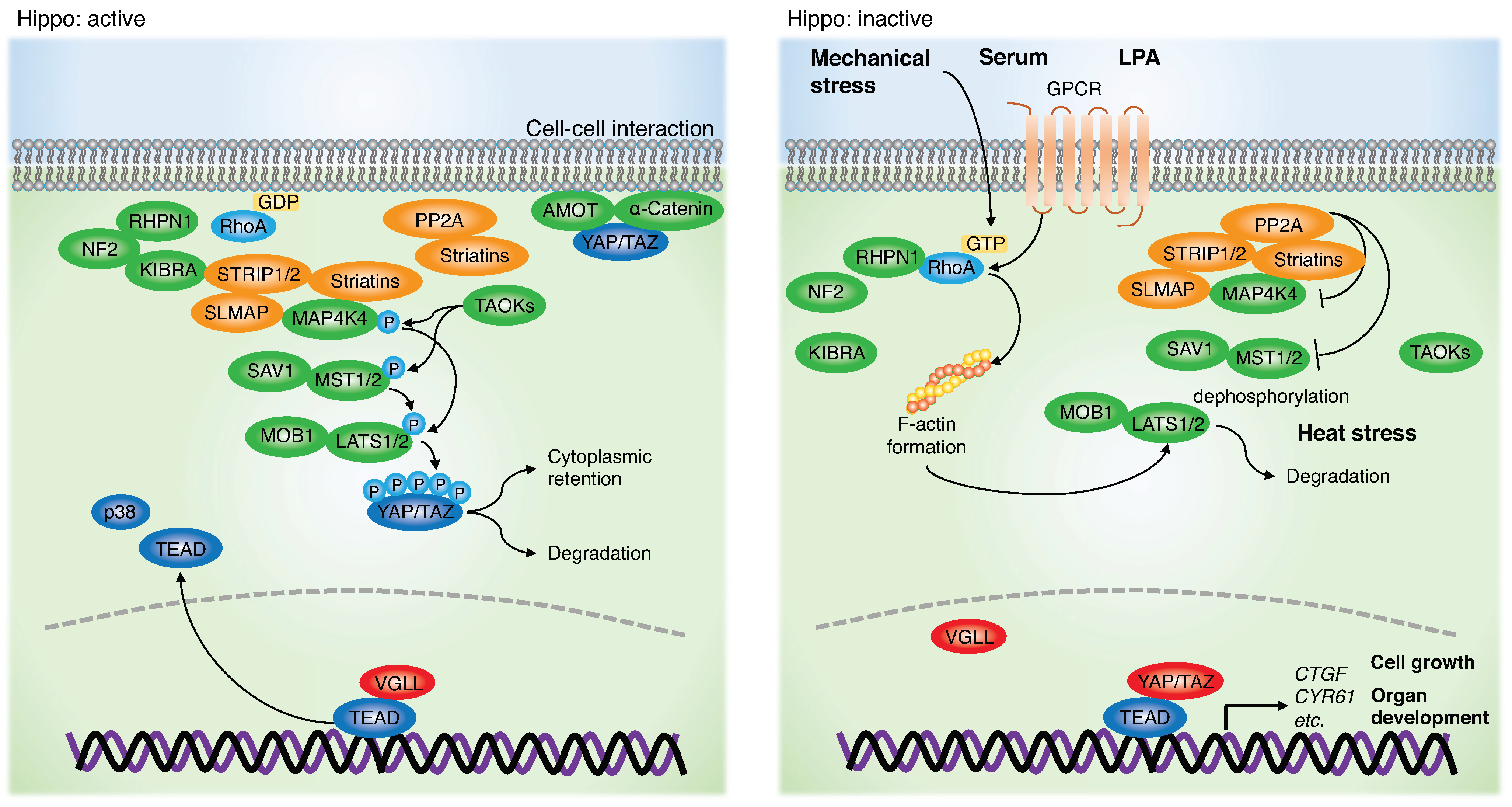
2. Role of the Hippo Pathway and YAP/TAZ in HNSCC
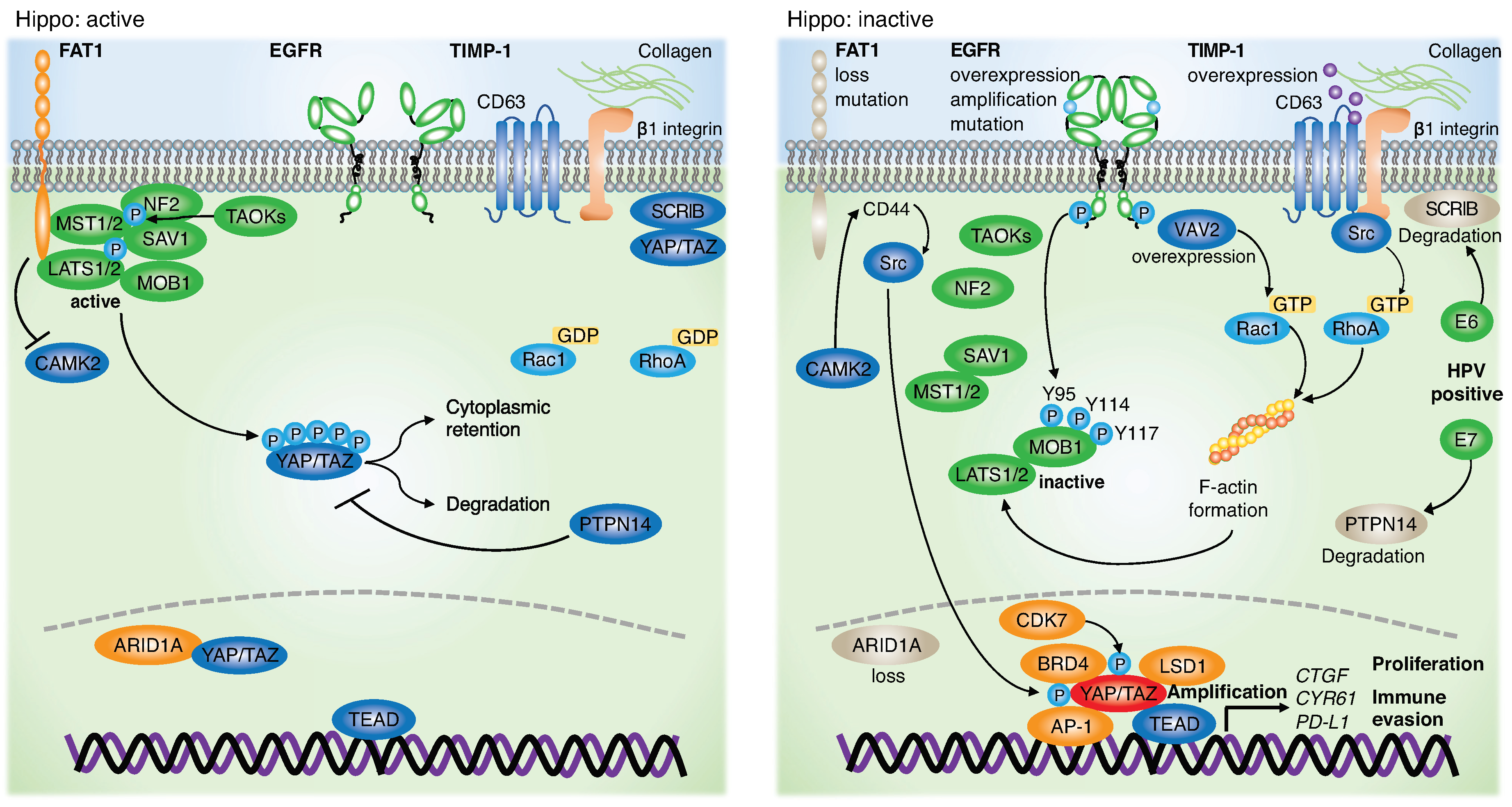
3. Genetic Alterations and Dysregulation of the Hippo Pathway in HNSCC
4. Therapeutic Approach Targeting the Hippo Pathway and YAP/TAZ
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Pfleger, C.M.; Hariharan, I.K. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003, 114, 457–467. [Google Scholar] [CrossRef]
- Udan, R.S.; Kango-Singh, M.; Nolo, R.; Tao, C.; Halder, G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 2003, 5, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Pantalacci, S.; Tapon, N.; Léopold, P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 2003, 5, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.; Schiripo, T.A.; Haber, D.A.; Hariharan, I.K. salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef]
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying tumor suppressors in genetic mosaics: The Drosophila lats gene encodes a putative protein kinase. Development 1995, 121, 1053–1063. [Google Scholar] [CrossRef]
- Kango-Singh, M.; Nolo, R.; Tao, C.; Verstreken, P.; Hiesinger, P.R.; Bellen, H.J.; Halder, G. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development 2002, 129, 5719–5730. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Mullett, S.J.; Stewart, A.F. Vgl-4, a novel member of the vestigial-like family of transcription cofactors, regulates alpha1-adrenergic activation of gene expression in cardiac myocytes. J. Biol. Chem. 2004, 279, 30800–30806. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Chapman, D.L.; Stewart, A.F. Mammalian vestigial-like 2, a cofactor of TEF-1 and MEF2 transcription factors that promotes skeletal muscle differentiation. J. Biol. Chem. 2002, 277, 48889–48898. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Chan, S.W.; Lee, I.; Song, H.; Hong, W. Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure 2012, 20, 1135–1140. [Google Scholar] [CrossRef]
- Vaudin, P.; Delanoue, R.; Davidson, I.; Silber, J.; Zider, A. TONDU (TDU), a novel human protein related to the product of vestigial (vg) gene of Drosophila melanogaster interacts with vertebrate TEF factors and substitutes for Vg function in wing formation. Development 1999, 126, 4807–4816. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Lu, Y.; Li, P.; Yin, M.X.; Lv, D.; Zhang, W.; Wang, H.; Zhou, Z.; Ji, H.; Zhao, Y.; et al. A novel partner of Scalloped regulates Hippo signaling via antagonizing Scalloped-Yorkie activity. Cell Res. 2013, 23, 1201–1214. [Google Scholar] [CrossRef]
- Koontz, L.M.; Liu-Chittenden, Y.; Yin, F.; Zheng, Y.; Yu, J.; Huang, B.; Chen, Q.; Wu, S.; Pan, D. The Hippo effector Yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev. Cell 2013, 25, 388–401. [Google Scholar] [CrossRef]
- Zhang, W.; Gao, Y.; Li, P.; Shi, Z.; Guo, T.; Li, F.; Han, X.; Feng, Y.; Zheng, C.; Wang, Z.; et al. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 2014, 24, 331–343. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, H.; Withers, H.G.; Yang, N.; Denson, K.E.; Mussell, A.L.; Truskinovsky, A.; Fan, Q.; Gelman, I.H.; Frangou, C.; et al. VGLL4 Selectively Represses YAP-Dependent Gene Induction and Tumorigenic Phenotypes in Breast Cancer. Sci. Rep. 2017, 7, 6190. [Google Scholar] [CrossRef]
- Moya, I.M.; Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plouffe, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.S.; Lu, W.; Lu, S.; et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 2015, 6, 8357. [Google Scholar] [CrossRef] [PubMed]
- Boggiano, J.C.; Vanderzalm, P.J.; Fehon, R.G. Tao-1 phosphorylates Hippo/MST kinases to regulate the Hippo-Salvador-Warts tumor suppressor pathway. Dev. Cell 2011, 21, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.L.; Lin, J.I.; Zhang, X.; Harvey, K.F. The sterile 20-like kinase Tao-1 controls tissue growth by regulating the Salvador-Warts-Hippo pathway. Dev. Cell 2011, 21, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Nirala, N.K.; Nie, Y.; Chen, H.J.; Ostroff, G.; Mao, J.; Wang, Q.; Xu, L.; Ip, Y.T. Ingestion of Food Particles Regulates the Mechanosensing Misshapen-Yorkie Pathway in Drosophila Intestinal Growth. Dev. Cell 2018, 45, 433–449.e6. [Google Scholar] [CrossRef]
- Shi, Z.; Jiao, S.; Zhou, Z. STRIPAK complexes in cell signaling and cancer. Oncogene 2016, 35, 4549–4557. [Google Scholar] [CrossRef]
- Chen, R.; Xie, R.; Meng, Z.; Ma, S.; Guan, K.L. STRIPAK integrates upstream signals to initiate the Hippo kinase cascade. Nat. Cell Biol. 2019, 21, 1565–1577. [Google Scholar] [CrossRef]
- Kim, J.W.; Berrios, C.; Kim, M.; Schade, A.E.; Adelmant, G.; Yeerna, H.; Damato, E.; Iniguez, A.B.; Florens, L.; Washburn, M.P.; et al. STRIPAK directs PP2A activity toward MAP4K4 to promote oncogenic transformation of human cells. Elife 2020, 9, e53003. [Google Scholar] [CrossRef]
- Luo, M.; Meng, Z.; Moroishi, T.; Lin, K.C.; Shen, G.; Mo, F.; Shao, B.; Wei, X.; Zhang, P.; Wei, Y.; et al. Heat stress activates YAP/TAZ to induce the heat shock transcriptome. Nat. Cell Biol. 2020, 22, 1447–1459. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, M.; Sun, J.; Lau, A.; Curtis, S.; Goldsmith, J.; Fox, V.L.; Wei, C.; Frazier, M.; Samson, O.; Wong, K.K.; et al. A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat. Cell Biol. 2014, 16, 108–117. [Google Scholar] [CrossRef]
- Pearson, J.D.; Huang, K.; Pacal, M.; McCurdy, S.R.; Lu, S.; Aubry, A.; Yu, T.; Wadosky, K.M.; Zhang, L.; Wang, T.; et al. Binary pan-cancer classes with distinct vulnerabilities defined by pro- or anti-cancer YAP/TEAD activity. Cancer Cell 2021, 39, 1115–1134.e12. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Wu, Z.; Yang, F.; Zhang, J.; Johnson, R.L.; Rosenfeld, M.G.; Guan, K.L. Hippo signalling maintains ER expression and ER+ breast cancer growth. Nature 2021, 591, E1–E10. [Google Scholar] [CrossRef]
- Ma, S.; Tang, T.; Probst, G.; Konradi, A.; Jin, C.; Li, F.; Silvio Gutkind, J.; Fu, X.D.; Guan, K.L. Transcriptional repression of estrogen receptor alpha by YAP reveals the Hippo pathway as therapeutic target for ER+ breast cancer. Nat. Commun. 2022, 13, 1061. [Google Scholar] [CrossRef]
- Cheung, P.; Xiol, J.; Dill, M.T.; Yuan, W.C.; Panero, R.; Roper, J.; Osorio, F.G.; Maglic, D.; Li, Q.; Gurung, B.; et al. Regenerative Reprogramming of the Intestinal Stem Cell State via Hippo Signaling Suppresses Metastatic Colorectal Cancer. Cell Stem Cell 2020, 27, 590–604.e9. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.; Kim, J. The potential role of YAP in head and neck squamous cell carcinoma. Exp. Mol. Med. 2020, 52, 1264–1274. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Degese, M.S.; Vitale-Cross, L.; Iglesias-Bartolome, R.; Valera, J.L.C.; Wang, Z.; Feng, X.; Yeerna, H.; Vadmal, V.; Moroishi, T.; et al. Assembly and activation of the Hippo signalome by FAT1 tumor suppressor. Nat. Commun. 2018, 9, 2372. [Google Scholar] [CrossRef] [PubMed]
- Omori, H.; Nishio, M.; Masuda, M.; Miyachi, Y.; Ueda, F.; Nakano, T.; Sato, K.; Mimori, K.; Taguchi, K.; Hikasa, H.; et al. YAP1 is a potent driver of the onset and progression of oral squamous cell carcinoma. Sci. Adv. 2020, 6, eaay3324. [Google Scholar] [CrossRef] [PubMed]
- Debaugnies, M.; Sánchez-Danés, A.; Rorive, S.; Raphaël, M.; Liagre, M.; Parent, M.A.; Brisebarre, A.; Salmon, I.; Blanpain, C. YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep. 2018, 19, e45809. [Google Scholar] [CrossRef]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.R.; et al. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef]
- Iglesias-Bartolome, R.; Torres, D.; Marone, R.; Feng, X.; Martin, D.; Simaan, M.; Chen, M.; Weinstein, L.S.; Taylor, S.S.; Molinolo, A.A.; et al. Inactivation of a Gα(s)-PKA tumour suppressor pathway in skin stem cells initiates basal-cell carcinogenesis. Nat. Cell Biol. 2015, 17, 793–803. [Google Scholar] [CrossRef]
- Ge, L.; Smail, M.; Meng, W.; Shyr, Y.; Ye, F.; Fan, K.H.; Li, X.; Zhou, H.M.; Bhowmick, N.A. Yes-associated protein expression in head and neck squamous cell carcinoma nodal metastasis. PLoS ONE 2011, 6, e27529. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef]
- Pal, A.; Barrett, T.F.; Paolini, R.; Parikh, A.; Puram, S.V. Partial EMT in head and neck cancer biology: A spectrum instead of a switch. Oncogene 2021, 40, 5049–5065. [Google Scholar] [CrossRef]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed]
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef]
- Callejas-Valera, J.L.; Iglesias-Bartolome, R.; Amornphimoltham, P.; Palacios-Garcia, J.; Martin, D.; Califano, J.A.; Molinolo, A.A.; Gutkind, J.S. mTOR inhibition prevents rapid-onset of carcinogen-induced malignancies in a novel inducible HPV-16 E6/E7 mouse model. Carcinogenesis 2016, 37, 1014–1025. [Google Scholar] [CrossRef]
- Wang, W.; Huang, J.; Wang, X.; Yuan, J.; Li, X.; Feng, L.; Park, J.I.; Chen, J. PTPN14 is required for the density-dependent control of YAP1. Genes Dev. 2012, 26, 1959–1971. [Google Scholar] [CrossRef]
- Poernbacher, I.; Baumgartner, R.; Marada, S.K.; Edwards, K.; Stocker, H. Drosophila Pez acts in Hippo signaling to restrict intestinal stem cell proliferation. Curr. Biol. 2012, 22, 389–396. [Google Scholar] [CrossRef]
- Mello, S.S.; Valente, L.J.; Raj, N.; Seoane, J.A.; Flowers, B.M.; McClendon, J.; Bieging-Rolett, K.T.; Lee, J.; Ivanochko, D.; Kozak, M.M.; et al. A p53 Super-tumor Suppressor Reveals a Tumor Suppressive p53-Ptpn14-Yap Axis in Pancreatic Cancer. Cancer Cell 2017, 32, 460–473.e6. [Google Scholar] [CrossRef]
- Knight, J.F.; Sung, V.Y.C.; Kuzmin, E.; Couzens, A.L.; de Verteuil, D.A.; Ratcliffe, C.D.H.; Coelho, P.P.; Johnson, R.M.; Samavarchi-Tehrani, P.; Gruosso, T.; et al. KIBRA (WWC1) Is a Metastasis Suppressor Gene Affected by Chromosome 5q Loss in Triple-Negative Breast Cancer. Cell Rep. 2018, 22, 3191–3205. [Google Scholar] [CrossRef] [Green Version]
- Hatterschide, J.; Castagnino, P.; Kim, H.W.; Sperry, S.M.; Montone, K.T.; Basu, D.; White, E.A. YAP1 activation by human papillomavirus E7 promotes basal cell identity in squamous epithelia. elife 2022, 11, e75466. [Google Scholar] [CrossRef] [PubMed]
- Messa, L.; Celegato, M.; Bertagnin, C.; Mercorelli, B.; Alvisi, G.; Banks, L.; Palù, G.; Loregian, A. The Dimeric Form of HPV16 E6 Is Crucial to Drive YAP/TAZ Upregulation through the Targeting of hScrib. Cancers 2021, 13, 4083. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Mao, D.; Hua, G.; Lv, X.; Chen, X.; Angeletti, P.C.; Dong, J.; Remmenga, S.W.; Rodabaugh, K.J.; Zhou, J.; et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med. 2015, 7, 1426–1449. [Google Scholar] [CrossRef] [PubMed]
- Webb Strickland, S.; Brimer, N.; Lyons, C.; Vande Pol, S.B. Human Papillomavirus E6 interaction with cellular PDZ domain proteins modulates YAP nuclear localization. Virology 2018, 516, 127–138. [Google Scholar] [CrossRef]
- Dunne, J.; Hanby, A.M.; Poulsom, R.; Jones, T.A.; Sheer, D.; Chin, W.G.; Da, S.M.; Zhao, Q.; Beverley, P.C.; Owen, M.J. Molecular cloning and tissue expression of FAT, the human homologue of the Drosophila fat gene that is located on chromosome 4q34-q35 and encodes a putative adhesion molecule. Genomics 1995, 30, 207–223. [Google Scholar] [CrossRef]
- Magg, T.; Schreiner, D.; Solis, G.P.; Bade, E.G.; Hofer, H.W. Processing of the human protocadherin Fat1 and translocation of its cytoplasmic domain to the nucleus. Exp. Cell Res. 2005, 307, 100–108. [Google Scholar] [CrossRef]
- Morris, L.G.; Kaufman, A.M.; Gong, Y.; Ramaswami, D.; Walsh, L.A.; Turcan, Ş.; Eng, S.; Kannan, K.; Zou, Y.; Peng, L.; et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013, 45, 253–261. [Google Scholar] [CrossRef]
- Lin, S.C.; Lin, L.H.; Yu, S.Y.; Kao, S.Y.; Chang, K.W.; Cheng, H.W.; Liu, C.J. FAT1 somatic mutations in head and neck carcinoma are associated with tumor progression and survival. Carcinogenesis 2018, 39, 1320–1330. [Google Scholar] [CrossRef]
- Huang, C.; Chen, L.; Savage, S.R.; Eguez, R.V.; Dou, Y.; Li, Y.; da Veiga Leprevost, F.; Jaehnig, E.J.; Lei, J.T.; Wen, B.; et al. Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 2021, 39, 361–379.e16. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, C.; Chen, J.; Wang, D.; Tu, J.; Van Waes, C.; Saba, N.F.; Chen, Z.G.; Chen, Z. The Proteomic Landscape of Growth Factor Signaling Networks Associated with FAT1 Mutations in Head and Neck Cancers. Cancer Res. 2021, 81, 4402–4416. [Google Scholar] [CrossRef]
- Pastushenko, I.; Mauri, F.; Song, Y.; de Cock, F.; Meeusen, B.; Swedlund, B.; Impens, F.; Van Haver, D.; Opitz, M.; Thery, M.; et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 2021, 589, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Xia, H.; Dai, X.; Yu, H.; Zhou, S.; Fan, Z.; Wei, G.; Tang, Q.; Gong, Q.; Bi, F. EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: The mechanism and its implications in targeted therapy. Cell Death Dis. 2018, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Kim, N.G.; Gumbiner, B.M. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl. Acad. Sci. USA 2013, 110, 2569–2574. [Google Scholar] [CrossRef]
- Zhang, Z.; Du, J.; Wang, S.; Shao, L.; Jin, K.; Li, F.; Wei, B.; Ding, W.; Fu, P.; van Dam, H.; et al. OTUB2 Promotes Cancer Metastasis via Hippo-Independent Activation of YAP and TAZ. Mol. Cell 2019, 73, 7–21.e7. [Google Scholar] [CrossRef]
- Ando, T.; Arang, N.; Wang, Z.; Costea, D.E.; Feng, X.; Goto, Y.; Izumi, H.; Gilardi, M.; Ando, K.; Gutkind, J.S. EGFR Regulates the Hippo pathway by promoting the tyrosine phosphorylation of MOB1. Commun. Biol. 2021, 4, 1237. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [Green Version]
- Byeon, H.K.; Ku, M.; Yang, J. Beyond EGFR inhibition: Multilateral combat strategies to stop the progression of head and neck cancer. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jerhammar, F.; Johansson, A.C.; Ceder, R.; Welander, J.; Jansson, A.; Grafström, R.C.; Söderkvist, P.; Roberg, K. YAP1 is a potential biomarker for cetuximab resistance in head and neck cancer. Oral Oncol. 2014, 50, 832–839. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, Q.; Zhang, Q.; Li, Z.; Wang, E.; Qiu, X. Overexpression of yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci. 2010, 101, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.C.; You, B.; Yang, Y.L.; Zhang, W.Q.; Wang, Y.C.; Xu, Z.; Dai, Y.; Liu, S.; Yang, C.T.; Li, H.; et al. YAP promotes erlotinib resistance in human non-small cell lung cancer cells. Oncotarget 2016, 7, 51922–51933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, J.Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Meng, Z.; Qiu, Y.; Lin, K.C.; Kumar, A.; Placone, J.K.; Fang, C.; Wang, K.C.; Lu, S.; Pan, M.; Hong, A.W.; et al. RAP2 mediates mechanoresponses of the Hippo pathway. Nature 2018, 560, 655–660. [Google Scholar] [CrossRef]
- Cai, M.; Zheng, Z.; Bai, Z.; Ouyang, K.; Wu, Q.; Xu, S.; Huang, L.; Jiang, Y.; Wang, L.; Gao, J.; et al. Overexpression of angiogenic factors and matrix metalloproteinases in the saliva of oral squamous cell carcinoma patients: Potential non-invasive diagnostic and therapeutic biomarkers. BMC Cancer 2022, 22, 530. [Google Scholar] [CrossRef]
- Xia, Y.; Yeddula, N.; Leblanc, M.; Ke, E.; Zhang, Y.; Oldfield, E.; Shaw, R.J.; Verma, I.M. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat. Cell Biol. 2012, 14, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Toricelli, M.; Melo, F.H.; Peres, G.B.; Silva, D.C.; Jasiulionis, M.G. Timp1 interacts with beta-1 integrin and CD63 along melanoma genesis and confers anoikis resistance by activating PI3-K signaling pathway independently of Akt phosphorylation. Mol. Cancer 2013, 12, 1095. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Xu, S.; Zhang, H.; Wang, Y.; Xiao, C.; Jiang, T.; Wu, L.; Zhang, T.; Sun, X.; Zhong, L.; et al. TIMP1 is a prognostic marker for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway. J. Exp. Clin. Cancer Res. 2016, 35, 148. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.K.; Liu, X.W.; Chirco, R.; Fridman, R.; Kim, H.R. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 2006, 25, 3934–3942. [Google Scholar] [CrossRef] [PubMed]
- Chirco, R.; Liu, X.W.; Jung, K.K.; Kim, H.R. Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev. 2006, 25, 99–113. [Google Scholar] [CrossRef]
- Ando, T.; Charindra, D.; Shrestha, M.; Umehara, H.; Ogawa, I.; Miyauchi, M.; Takata, T. Tissue inhibitor of metalloproteinase-1 promotes cell proliferation through YAP/TAZ activation in cancer. Oncogene 2018, 37, 263–270. [Google Scholar] [CrossRef]
- Shrestha, M.; Ando, T.; Chea, C.; Sakamoto, S.; Nishisaka, T.; Ogawa, I.; Miyauchi, M.; Takata, T. The transition of tissue inhibitor of metalloproteinases from −4 to −1 induces aggressive behavior and poor patient survival in dedifferentiated liposarcoma via YAP/TAZ activation. Carcinogenesis 2019, 40, 1288–1297. [Google Scholar] [CrossRef]
- Gong, Y.; Scott, E.; Lu, R.; Xu, Y.; Oh, W.K.; Yu, Q. TIMP-1 promotes accumulation of cancer associated fibroblasts and cancer progression. PLoS ONE 2013, 8, e77366. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Rodríguez-Fdez, S.; Bustelo, X.R. The Vav GEF Family: An Evolutionary and Functional Perspective. Cells 2019, 8, 465. [Google Scholar] [CrossRef]
- Patel, V.; Rosenfeldt, H.M.; Lyons, R.; Servitja, J.M.; Bustelo, X.R.; Siroff, M.; Gutkind, J.S. Persistent activation of Rac1 in squamous carcinomas of the head and neck: Evidence for an EGFR/Vav2 signaling axis involved in cell invasion. Carcinogenesis 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo-Martín, L.F.; Fernández-Parejo, N.; Menacho-Márquez, M.; Rodríguez-Fdez, S.; Robles-Valero, J.; Zumalave, S.; Fabbiano, S.; Pascual, G.; García-Pedrero, J.M.; Abad, A.; et al. VAV2 signaling promotes regenerative proliferation in both cutaneous and head and neck squamous cell carcinoma. Nat. Commun. 2020, 11, 4788. [Google Scholar] [CrossRef] [PubMed]
- Thalappilly, S.; Soubeyran, P.; Iovanna, J.L.; Dusetti, N.J. VAV2 regulates epidermal growth factor receptor endocytosis and degradation. Oncogene 2010, 29, 2528–2539. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Koo, J.H.; Plouffe, S.W.; Meng, Z.; Lee, D.H.; Yang, D.; Lim, D.S.; Wang, C.Y.; Guan, K.L. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev. 2020, 34, 72–86. [Google Scholar] [CrossRef]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Galli, G.G.; Carrara, M.; Yuan, W.C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef]
- Cho, Y.S.; Li, S.; Wang, X.; Zhu, J.; Zhuo, S.; Han, Y.; Yue, T.; Yang, Y.; Jiang, J. CDK7 regulates organ size and tumor growth by safeguarding the Hippo pathway effector Yki/Yap/Taz in the nucleus. Genes Dev. 2020, 34, 53–71. [Google Scholar] [CrossRef]
- Alhousami, T.; Diny, M.; Ali, F.; Shin, J.; Kumar, G.; Kumar, V.; Campbell, J.D.; Noonan, V.; Hanna, G.J.; Denis, G.V.; et al. Inhibition of LSD1 Attenuates Oral Cancer Development and Promotes Therapeutic Efficacy of Immune Checkpoint Blockade and YAP/TAZ Inhibition. Mol. Cancer Res. 2022, 20, 712–721. [Google Scholar] [CrossRef]
- Li, H.; Wu, B.K.; Kanchwala, M.; Cai, J.; Wang, L.; Xing, C.; Zheng, Y.; Pan, D. YAP/TAZ drives cell proliferation and tumour growth via a polyamine-eIF5A hypusination-LSD1 axis. Nat. Cell Biol. 2022, 24, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Saladi, S.V.; Ross, K.; Karaayvaz, M.; Tata, P.R.; Mou, H.; Rajagopal, J.; Ramaswamy, S.; Ellisen, L.W. ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell 2017, 31, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Park, D.I.; Lee, D.H.; Lee, J.E.; Yeo, M.K.; Park, Y.H.; Lim, D.S.; Choi, W.; Lee, D.H.; Yoo, G.; et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2017, 491, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Janse van Rensburg, H.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef]
- Miao, J.; Hsu, P.C.; Yang, Y.L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef]
- Hsu, P.C.; Miao, J.; Wang, Y.C.; Zhang, W.Q.; Yang, Y.L.; Wang, C.W.; Yang, C.T.; Huang, Z.; You, J.; Xu, Z.; et al. Inhibition of yes-associated protein down-regulates PD-L1 (CD274) expression in human malignant pleural mesothelioma. J. Cell Mol. Med. 2018, 22, 3139–3148. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, C.G.; Kim, S.K.; Shin, S.J.; Choe, E.A.; Park, S.H.; Shin, E.C.; Kim, J. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol. Res. 2018, 6, 255–266. [Google Scholar] [CrossRef]
- Chen, N.; Golczer, G.; Ghose, S.; Lin, B.; Langenbucher, A.; Webb, J.; Bhanot, H.; Abt, N.B.; Lin, D.; Varvares, M.; et al. YAP1 maintains active chromatin state in head and neck squamous cell carcinomas that promotes tumorigenesis through cooperation with BRD4. Cell Rep. 2022, 39, 110970. [Google Scholar] [CrossRef]
- Moroishi, T.; Hayashi, T.; Pan, W.W.; Fujita, Y.; Holt, M.V.; Qin, J.; Carson, D.A.; Guan, K.L. The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 2016, 167, 1525–1539.e17. [Google Scholar] [CrossRef]
- Macleod, A.R. Abstract ND11: The discovery and characterization of ION-537: A next generation antisense oligonucleotide inhibitor of YAP1 in preclinical cancer models. Cancer Res. 2021, 81, ND11. [Google Scholar] [CrossRef]
- Lin, K.C.; Moroishi, T.; Meng, Z.; Jeong, H.S.; Plouffe, S.W.; Sekido, Y.; Han, J.; Park, H.W.; Guan, K.L. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat. Cell Biol. 2017, 19, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Park, J.; Feng, A.; Awasthi, P.; Wang, Z.; Chen, Q.; Iglesias-Bartolome, R. YAP1/TAZ-TEAD transcriptional networks maintain skin homeostasis by regulating cell proliferation and limiting KLF4 activity. Nat. Commun. 2020, 11, 1472. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Han, X.; Hung, A.W.; Weiguang, S.; Huda, N.; Chen, G.Y.; Kang, C.; Chia, C.S.; Luo, X.; Hong, W.; et al. Targeting the Central Pocket in Human Transcription Factor TEAD as a Potential Cancer Therapeutic Strategy. Structure 2015, 23, 2076–2086. [Google Scholar] [CrossRef]
- Noland, C.L.; Gierke, S.; Schnier, P.D.; Murray, J.; Sandoval, W.N.; Sagolla, M.; Dey, A.; Hannoush, R.N.; Fairbrother, W.J.; Cunningham, C.N. Palmitoylation of TEAD Transcription Factors Is Required for Their Stability and Function in Hippo Pathway Signaling. Structure 2016, 24, 179–186. [Google Scholar] [CrossRef]
- Chan, P.; Han, X.; Zheng, B.; DeRan, M.; Yu, J.; Jarugumilli, G.K.; Deng, H.; Pan, D.; Luo, X.; Wu, X. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat. Chem. Biol. 2016, 12, 282–289. [Google Scholar] [CrossRef]
- Kim, N.G.; Gumbiner, B.M. Cell contact and Nf2/Merlin-dependent regulation of TEAD palmitoylation and activity. Proc. Natl. Acad. Sci. USA 2019, 116, 9877–9882. [Google Scholar] [CrossRef]
- Lu, W.; Wang, J.; Li, Y.; Tao, H.; Xiong, H.; Lian, F.; Gao, J.; Ma, H.; Lu, T.; Zhang, D.; et al. Discovery and biological evaluation of vinylsulfonamide derivatives as highly potent, covalent TEAD autopalmitoylation inhibitors. Eur. J. Med. Chem. 2019, 184, 111767. [Google Scholar] [CrossRef]
- Li, Q.; Sun, Y.; Jarugumilli, G.K.; Liu, S.; Dang, K.; Cotton, J.L.; Xiol, J.; Chan, P.Y.; DeRan, M.; Ma, L.; et al. Lats1/2 Sustain Intestinal Stem Cells and Wnt Activation through TEAD-Dependent and Independent Transcription. Cell Stem. Cell 2020, 26, 675–692.e8. [Google Scholar] [CrossRef]
- Holden, J.K.; Crawford, J.J.; Noland, C.L.; Schmidt, S.; Zbieg, J.R.; Lacap, J.A.; Zang, R.; Miller, G.M.; Zhang, Y.; Beroza, P.; et al. Small Molecule Dysregulation of TEAD Lipidation Induces a Dominant-Negative Inhibition of Hippo Pathway Signaling. Cell Rep. 2020, 31, 107809. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.T.; Konradi, A.W.; Feng, Y.; Peng, X.; Ma, M.; Li, J.; Yu, F.X.; Guan, K.L.; Post, L. Small Molecule Inhibitors of TEAD Auto-palmitoylation Selectively Inhibit Proliferation and Tumor Growth of NF2-deficient Mesothelioma. Mol. Cancer Ther. 2021, 20, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e12. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Lakhani, N.J.; McKean, M.; Lingaraj, T.; Victor, L.; Sanchez-Martin, M.; Kacena, K.; Malek, K.S.; Santillana, S. A phase 1, first-in-human study of IK-930, an oral TEAD inhibitor targeting the Hippo pathway in subjects with advanced solid tumors. J. Clin. Oncol. 2022, 40, TPS3168. [Google Scholar] [CrossRef]
- Esposito, D.; Pant, I.; Shen, Y.; Qiao, R.F.; Yang, X.; Bai, Y.; Jin, J.; Poulikakos, P.I.; Aaronson, S.A. ROCK1 mechano-signaling dependency of human malignancies driven by TEAD/YAP activation. Nat. Commun. 2022, 13, 703. [Google Scholar] [CrossRef]
- Mudianto, T.; Campbell, K.M.; Webb, J.; Zolkind, P.; Skidmore, Z.L.; Riley, R.; Barnell, E.K.; Ozgenc, I.; Giri, T.; Dunn, G.P.; et al. Yap1 Mediates Trametinib Resistance in Head and Neck Squamous Cell Carcinomas. Clin. Cancer Res. 2021, 27, 2326–2339. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef]
- Taniguchi, K.; Wu, L.W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef]
- Feng, X.; Arang, N.; Rigiracciolo, D.C.; Lee, J.S.; Yeerna, H.; Wang, Z.; Lubrano, S.; Kishore, A.; Pachter, J.A.; König, G.M.; et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 2019, 35, 457–472.e5. [Google Scholar] [CrossRef]
- Goto, Y.; Ando, T.; Izumi, H.; Feng, X.; Arang, N.; Gilardi, M.; Wang, Z.; Ando, K.; Gutkind, J.S. Muscarinic receptors promote castration-resistant growth of prostate cancer through a FAK-YAP signaling axis. Oncogene 2020, 39, 4014–4027. [Google Scholar] [CrossRef]
- Li, P.; Silvis, M.R.; Honaker, Y.; Lien, W.H.; Arron, S.T.; Vasioukhin, V. αE-catenin inhibits a Src-YAP1 oncogenic module that couples tyrosine kinases and the effector of Hippo signaling pathway. Genes Dev. 2016, 30, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.S.; Guan, K.L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed]
- DeRan, M.; Yang, J.; Shen, C.H.; Peters, E.C.; Fitamant, J.; Chan, P.; Hsieh, M.; Zhu, S.; Asara, J.M.; Zheng, B.; et al. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 2014, 9, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zhu, Y.; Zhang, W.; Liao, Q.; Chen, Y.; Zhao, X.; Guo, Q.; Shen, P.; Zhen, B.; Qian, X.; et al. Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Mol. Cell 2017, 68, 591–604.e5. [Google Scholar] [CrossRef]
- Wu, X.; Yeerna, H.; Goto, Y.; Ando, T.; Wu, V.H.; Zhang, X.; Wang, Z.; Amornphimoltham, P.; Murphy, A.N.; Tamayo, P.; et al. Metformin Inhibits Progression of Head and Neck Squamous Cell Carcinoma by Acting Directly on Carcinoma-Initiating Cells. Cancer Res. 2019, 79, 4360–4370. [Google Scholar] [CrossRef]
- Gulati, S.; Desai, J.; Palackdharry, S.M.; Morris, J.C.; Zhu, Z.; Jandarov, R.; Riaz, M.K.; Takiar, V.; Mierzwa, M.; Gutkind, J.S.; et al. Phase 1 dose-finding study of metformin in combination with concurrent cisplatin and radiotherapy in patients with locally advanced head and neck squamous cell cancer. Cancer 2020, 126, 354–362. [Google Scholar] [CrossRef]
- Gutkind, J.S.; Molinolo, A.A.; Wu, X.; Wang, Z.; Nachmanson, D.; Harismendy, O.; Alexandrov, L.B.; Wuertz, B.R.; Ondrey, F.G.; Laronde, D.; et al. Inhibition of mTOR signaling and clinical activity of metformin in oral premalignant lesions. JCI Insight 2021, 6, e147096. [Google Scholar] [CrossRef]
- Vitale-Cross, L.; Molinolo, A.A.; Martin, D.; Younis, R.H.; Maruyama, T.; Patel, V.; Chen, W.; Schneider, A.; Gutkind, J.S. Metformin prevents the development of oral squamous cell carcinomas from carcinogen-induced premalignant lesions. Cancer Prev. Res. 2012, 5, 562–573. [Google Scholar] [CrossRef]
- Madera, D.; Vitale-Cross, L.; Martin, D.; Schneider, A.; Molinolo, A.A.; Gangane, N.; Carey, T.E.; McHugh, J.B.; Komarck, C.M.; Walline, H.M.; et al. Prevention of tumor growth driven by PIK3CA and HPV oncogenes by targeting mTOR signaling with metformin in oral squamous carcinomas expressing OCT3. Cancer Prev. Res. 2015, 8, 197–207. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Company | Target | Clinical Trials (ClinicalTrials.gov Identifier) | Cancer Type |
|---|---|---|---|---|
| IK-930 | Ikena Oncology | TEAD | Phase I (NCT05228015) | Solid, unspecified |
| ION-537 | Ionis Pharmacueticals | YAP | Phase I (NCT04659096) | Solid, unspecified |
| BTX-A51 | BioTheryX | CDK7 | Phase I (NCT04872166) | Solid, unspecified |
| fadraciclib | Cyclacel | CDK7 | Phase II (NCT04983810) | Solid, unspecified |
| samuraciclib | Carrick Therapeutics, Evotec | CDK7 | Phase II (NCT03363893) | Unspecified |
| SY-5609 | Syros Pharmaceuticals | CDK7 | Phase I (NCT04247126) | Solid, unspecified |
| XL-102 | Exelixis | CDK7 | Phase I (NCT04726332) | Solid, unspecified |
| trametinib | Novartis | MEK | Phase II (NCT01376310) | Solid, unspecified |
| binimetinib | Pfizer | MEK | Phase II (NCT01885195) | Solid, unspecified |
| cobimetinib | Roche | MEK | Phase II (NCT02639546) | Solid, unspecified |
| mirdametinib | SpringWorks Therapeutics | MEK | Phase II (NCT05054374) | Solid, unspecified |
| BI-894999 | Boehringer Ingelheim | BRD4 | Phase I (NCT02516553) | Solid, unspecified |
| BPI-23314 | Betta Pharmaceuticals | BRD4 | Phase I (CTR20192223, China FDA) | Solid, unspecified |
| NUV-868 | Nuvation Bio | BRD4 | Phase II (NCT05252390) | Solid, unspecified |
| PLX-2853 | Daiichi Sankyo | BRD4 | Phase II (NCT03297424) | Solid, unspecified |
| SYHA-1801 | CSPC Pharmaceutical | BRD4 | Phase I (NCT04309968) | Solid, unspecified |
| JBI-802 | Jubilant Life Sciences | LSD1 | Phase II (NCT05268666) | Solid, unspecified |
| seclidemstat | Salarius Pharmaceuticals | LSD1 | Phase II (NCT05266196) | Solid, unspecified |
| CC 90011 | Bristol-Myers Squibb | LSD1 | Phase II (NCT02875223) | Solid, unspecified |
| dasatinib | Bristol-Myers Squibb | Src | Phase II (NCT00882583) | Head and neck |
| repotrectinib | Turning Point Therapeutics, Zai Lab | Src | Phase III (NCT05004116) | Solid, unspecified |
| VAL-201 | ValiRx | Src | Phase II (NCT02280317) | Solid, unspecified |
| TPX-0046 | Turning Point Therapeutics | Src | Phase II (NCT04161391) | Solid, unspecified |
| elzovantinib | Turning Point Therapeutics | Src | Phase II (NCT03993873) | Solid, unspecified |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ando, T.; Okamoto, K.; Shintani, T.; Yanamoto, S.; Miyauchi, M.; Gutkind, J.S.; Kajiya, M. Integrating Genetic Alterations and the Hippo Pathway in Head and Neck Squamous Cell Carcinoma for Future Precision Medicine. J. Pers. Med. 2022, 12, 1544. https://doi.org/10.3390/jpm12101544
Ando T, Okamoto K, Shintani T, Yanamoto S, Miyauchi M, Gutkind JS, Kajiya M. Integrating Genetic Alterations and the Hippo Pathway in Head and Neck Squamous Cell Carcinoma for Future Precision Medicine. Journal of Personalized Medicine. 2022; 12(10):1544. https://doi.org/10.3390/jpm12101544
Chicago/Turabian StyleAndo, Toshinori, Kento Okamoto, Tomoaki Shintani, Souichi Yanamoto, Mutsumi Miyauchi, J. Silvio Gutkind, and Mikihito Kajiya. 2022. "Integrating Genetic Alterations and the Hippo Pathway in Head and Neck Squamous Cell Carcinoma for Future Precision Medicine" Journal of Personalized Medicine 12, no. 10: 1544. https://doi.org/10.3390/jpm12101544
APA StyleAndo, T., Okamoto, K., Shintani, T., Yanamoto, S., Miyauchi, M., Gutkind, J. S., & Kajiya, M. (2022). Integrating Genetic Alterations and the Hippo Pathway in Head and Neck Squamous Cell Carcinoma for Future Precision Medicine. Journal of Personalized Medicine, 12(10), 1544. https://doi.org/10.3390/jpm12101544