
Adipogenic Signaling Promotes Arrhythmia Substrates before Structural Abnormalities in TMEM43 ARVC


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Differentiation of Human Myocytes from iPSCs
2.2. Differentiation of MSCs from Human iPSCs
2.3. TMEM43 Expression Analysis by Quantitative Real-Time RT-PCR (qPCR)
2.4. DNA Extraction and TMEM43 Genotyping
2.5. TMEM43 Total Protein Analysis
2.6. Early Adipogenic Condition
2.7. ELISA and Surface Marker Expression
2.8. Optical Mapping and Action Potential Analysis
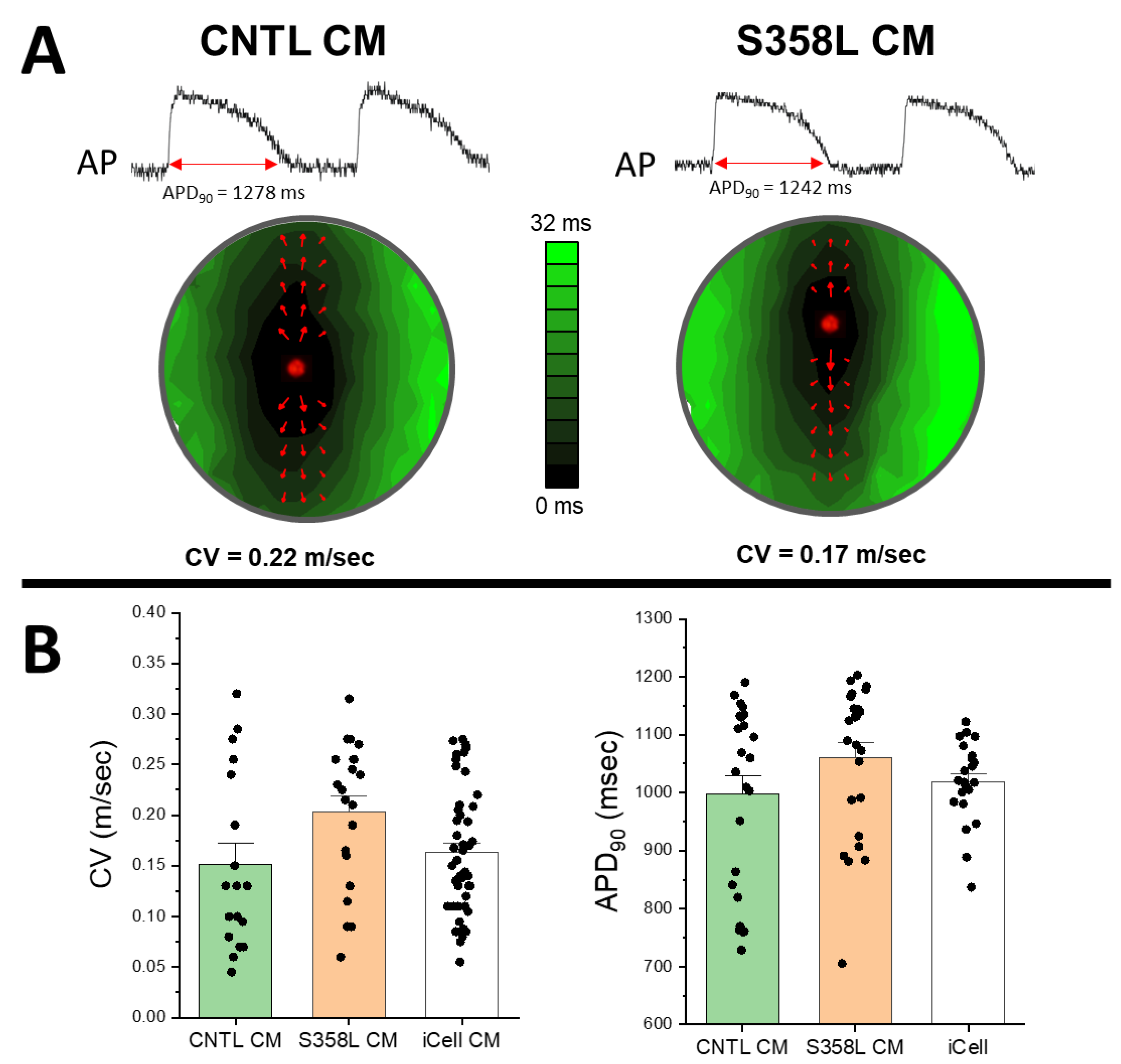
3. Results
4. Discussion
4.1. Early Arrhythmia Substrates Are Independent of the S358L Mutation in Myocytes
4.2. Adipogenic Conditions and Arrhythmia Substrates
4.3. The Role of IGF-1 in Early Arrhythmogenesis in ARVC
5. Limitations
6. Clinical Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Ingles, J.; Bagnall, R.D.; Yeates, L.; McGrady, M.; Berman, Y.; Whalley, D.; Duflou, J.; Semsarian, C. Concealed Arrhythmogenic Right Ventricular Cardiomyopathy in Sudden Unexplained Cardiac Death Events. Circ. Genom. Precis. Med. 2018, 11, e002355. [Google Scholar] [CrossRef]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J. Am. Coll. Cardiol. 2013, 62, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Calkins, H. Present understanding of the relationship between exercise and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Trends Cardiovasc. Med. 2015, 25, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Paulin, F.L.; Hodgkinson, K.A.; MacLaughlan, S.; Stuckless, S.N.; Templeton, C.; Shah, S.; Bremner, H.; Roberts, J.D.; Young, T.L.; Parfrey, P.S.; et al. Exercise and arrhythmic risk in TMEM43 p.S358L arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2020, 17, 1159–1166. [Google Scholar] [CrossRef]
- Mu, J.; Zhang, G.; Xue, D.; Xi, M.; Qi, J.; Dong, H. Sudden cardiac death owing to arrhythmogenic right ventricular cardiomyopathy: Two case reports and systematic literature review. Medicine 2017, 96, e8808. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Padron-Barthe, L.; Villalba-Orero, M.; Gomez-Salinero, J.M.; Dominguez, F.; Roman, M.; Larrasa-Alonso, J.; Ortiz-Sanchez, P.; Martinez, F.; Lopez-Olaneta, M.; Bonzon-Kulichenko, E.; et al. Severe Cardiac Dysfunction and Death Caused by Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Are Improved by Inhibition of Glycogen Synthase Kinase-3beta. Circulation 2019, 140, 1188–1204. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Djouadi, F.; Lecarpentier, Y.; Hebert, J.L.; Charron, P.; Bastin, J.; Coirault, C. A potential link between peroxisome proliferator-activated receptor signalling and the pathogenesis of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Res. 2009, 84, 83–90. [Google Scholar] [CrossRef]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wei, H.; Lu, J.; Ho, S.; Zhang, G.; Sun, X.; Oh, Y.; Tan, S.H.; Ng, M.L.; Shim, W.; et al. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 1122–1133. [Google Scholar] [CrossRef]
- Lombardi, R.; Chen, S.N.; Ruggiero, A.; Gurha, P.; Czernuszewicz, G.Z.; Willerson, J.T.; Marian, A.J. Cardiac Fibro-Adipocyte Progenitors Express Desmosome Proteins and Preferentially Differentiate to Adipocytes Upon Deletion of the Desmoplakin Gene. Circ. Res. 2016, 119, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Roderburg, C.; Wein, F.; Diehlmann, A.; Frankhauser, M.; Schubert, R.; Eckstein, V.; Ho, A.D. Molecular and secretory profiles of human mesenchymal stromal cells and their abilities to maintain primitive hematopoietic progenitors. Stem Cells 2007, 25, 2638–2647. [Google Scholar] [CrossRef] [PubMed]
- Sattayaprasert, P.; Vasireddi, S.K.; Bektik, E.; Jeon, O.; Hajjiri, M.; Mackall, J.A.; Moravec, C.S.; Alsberg, E.; Fu, J.; Laurita, K.R. Human Cardiac Mesenchymal Stem Cells Remodel in Disease and Can Regulate Arrhythmia Substrates. Circ. Arrhythm. Electrophysiol. 2020, 13, e008740. [Google Scholar] [CrossRef] [PubMed]
- Monsanto, M.M.; White, K.S.; Kim, T.; Wang, B.J.; Fisher, K.; Ilves, K.; Khalafalla, F.G.; Casillas, A.; Broughton, K.; Mohsin, S.; et al. Concurrent Isolation of Three Distinct Cardiac Stem Cell Populations from a Single Human Heart Biopsy. Circ. Res. 2017, 121, 113–124. [Google Scholar] [CrossRef]
- Scalco, A.; Liboni, C.; Angioni, R.; Di Bona, A.; Albiero, M.; Bertoldi, N.; Fadini, G.P.; Thiene, G.; Chelko, S.P.; Basso, C.; et al. Arrhythmogenic Cardiomyopathy Is a Multicellular Disease Affecting Cardiac and Bone Marrow Mesenchymal Stromal Cells. J. Clin. Med. 2021, 10, 1871. [Google Scholar] [CrossRef]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Dello Russo, A.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Heart J. 2016, 37, 1835–1846. [Google Scholar] [CrossRef]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef]
- Tohyama, S.; Fujita, J.; Fujita, C.; Yamaguchi, M.; Kanaami, S.; Ohno, R.; Sakamoto, K.; Kodama, M.; Kurokawa, J.; Kanazawa, H.; et al. Efficient Large-Scale 2D Culture System for Human Induced Pluripotent Stem Cells and Differentiated Cardiomyocytes. Stem Cell Rep. 2017, 9, 1406–1414. [Google Scholar] [CrossRef]
- Hynes, K.; Menicanin, D.; Mrozik, K.; Gronthos, S.; Bartold, P.M. Generation of functional mesenchymal stem cells from different induced pluripotent stem cell lines. Stem Cells Dev. 2014, 23, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Sattayaprasert, P.; Nassal, D.M.; Wan, X.; Deschenes, I.; Laurita, K.R. Mesenchymal stem cells suppress cardiac alternans by activation of PI3K mediated nitroso-redox pathway. J. Mol. Cell. Cardiol. 2016, 98, 138–145. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McPheeters, M.T.; Wang, Y.T.; Werdich, A.A.; Jenkins, M.W.; Laurita, K.R. An infrared optical pacing system for screening cardiac electrophysiology in human cardiomyocytes. PLoS ONE 2017, 12, e0183761. [Google Scholar] [CrossRef] [PubMed]
- Tosaki, A. ArrhythmoGenoPharmacoTherapy. Front. Pharmacol. 2020, 11, 616. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef]
- Yu, J.K.; Liang, J.A.; Franceschi, W.H.; Huang, Q.; Pashakhanloo, F.; Sung, E.; Boyle, P.M.; Trayanova, N.A. Assessment of arrhythmia mechanism and burden of the infarcted ventricles following remuscularization with pluripotent stem cell-derived cardiomyocyte patches using patient-derived models. Cardiovasc. Res. 2022, 118, 1247–1261. [Google Scholar] [CrossRef]
- Andrews, C.M.; Srinivasan, N.T.; Rosmini, S.; Bulluck, H.; Orini, M.; Jenkins, S.; Pantazis, A.; McKenna, W.J.; Moon, J.C.; Lambiase, P.D.; et al. Electrical and Structural Substrate of Arrhythmogenic Right Ventricular Cardiomyopathy Determined Using Noninvasive Electrocardiographic Imaging and Late Gadolinium Magnetic Resonance Imaging. Circ. Arrhythm. Electrophysiol. 2017, 10, e005105. [Google Scholar] [CrossRef]
- Cruz, F.M.; Sanz-Rosa, D.; Roche-Molina, M.; Garcia-Prieto, J.; Garcia-Ruiz, J.M.; Pizarro, G.; Jimenez-Borreguero, L.J.; Torres, M.; Bernad, A.; Ruiz-Cabello, J.; et al. Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J. Am. Coll. Cardiol. 2015, 65, 1438–1450. [Google Scholar] [CrossRef]
- Siragam, V.; Cui, X.; Masse, S.; Ackerley, C.; Aafaqi, S.; Strandberg, L.; Tropak, M.; Fridman, M.D.; Nanthakumar, K.; Liu, J.; et al. TMEM43 mutation p.S358L alters intercalated disc protein expression and reduces conduction velocity in arrhythmogenic right ventricular cardiomyopathy. PLoS ONE 2014, 9, e109128. [Google Scholar] [CrossRef]
- Lyon, R.C.; Mezzano, V.; Wright, A.T.; Pfeiffer, E.; Chuang, J.; Banares, K.; Castaneda, A.; Ouyang, K.; Cui, L.; Contu, R.; et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum. Mol. Genet. 2014, 23, 1134–1150. [Google Scholar] [CrossRef]
- Dou, W.; Zhao, Q.; Malhi, M.; Liu, X.; Zhang, Z.; Wang, L.; Masse, S.; Nanthakumar, K.; Hamilton, R.; Maynes, J.T.; et al. Label-free conduction velocity mapping and gap junction assessment of functional iPSC-Cardiomyocyte monolayers. Biosens. Bioelectron. 2020, 167, 112468. [Google Scholar] [CrossRef]
- Kim, J.C.; Perez-Hernandez, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca(2+)i Homeostasis and Connexin 43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2-Deficient Mice. Circulation 2019, 140, 1015–1030. [Google Scholar] [CrossRef]
- Rajkumar, R.; Sembrat, J.C.; McDonough, B.; Seidman, C.E.; Ahmad, F. Functional effects of the TMEM43 Ser358Leu mutation in the pathogenesis of arrhythmogenic right ventricular cardiomyopathy. BMC Med. Genet. 2012, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, R.; Fu, H.; Li, J.; Wang, X.; Cheng, L.; Korantzopoulos, P.; Tse, G.; Li, G.; Liu, T. Pioglitazone attenuates atrial remodeling and vulnerability to atrial fibrillation in alloxan-induced diabetic rabbits. Cardiovasc. Ther. 2017, 35, e12284. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wang, L.; Hou, J.; Guo, T.; Xing, Y.; Zheng, S.; Zhou, C.; Huang, H.; Long, H.; Zhong, T.; et al. Peroxisome Proliferator-Activated Receptor Gamma Promotes Mesenchymal Stem Cells to Express Connexin43 via the Inhibition of TGF-beta1/Smads Signaling in a Rat Model of Myocardial Infarction. Stem Cell Rev. Rep. 2015, 11, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.P.; Katchman, A.; Son, N.H.; Trent, C.M.; Khan, R.; Shiomi, T.; Huang, H.; Amin, V.; Lader, J.M.; Vasquez, C.; et al. Mice with cardiac overexpression of peroxisome proliferator-activated receptor gamma have impaired repolarization and spontaneous fatal ventricular arrhythmias. Circulation 2011, 124, 2812–2821. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of adipogenesis by Wnt signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; van den Hoff, M.J.; de Bakker, J.M.; et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Tandri, H.; Duffy, E.R.; Winterfield, J.R.; Mackey-Bojack, S.; Picken, M.M.; Cooper, L.T.; Wilber, D.J.; Marcus, F.I.; Basso, C.; et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2011, 4, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.Y.; et al. Evidence from Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy: Associations of Circulating Anti-Heart and Anti-Intercalated Disk Autoantibodies with Disease Severity and Family History. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Iekushi, K.; Seeger, F.; Assmus, B.; Zeiher, A.M.; Dimmeler, S. Regulation of cardiac microRNAs by bone marrow mononuclear cell therapy in myocardial infarction. Circulation 2012, 125, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Haider, H.K.H.; Jiang, S.; Idris, N.M.; Ashraf, M. IGF-1-overexpressing mesenchymal stem cells accelerate bone marrow stem cell mobilization via paracrine activation of SDF-1alpha/CXCR4 signaling to promote myocardial repair. Circ. Res. 2008, 103, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Mughal, A.; Kumar, D.; Vikram, A. Effects of Thiazolidinediones on metabolism and cancer: Relative influence of PPARgamma and IGF-1 signaling. Eur. J. Pharmacol. 2015, 768, 217–225. [Google Scholar] [CrossRef]
- Blazeski, A.; Lowenthal, J.; Wang, Y.; Teuben, R.; Zhu, R.; Gerecht, S.; Tomaselli, G.; Tung, L. Engineered Heart Slice Model of Arrhythmogenic Cardiomyopathy Using Plakophilin-2 Mutant Myocytes. Tissue Eng. Part A 2019, 25, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Sommariva, E.; Stadiotti, I.; Casella, M.; Catto, V.; Dello Russo, A.; Carbucicchio, C.; Arnaboldi, L.; De Metrio, S.; Milano, G.; Scopece, A.; et al. Oxidized LDL-dependent pathway as new pathogenic trigger in arrhythmogenic cardiomyopathy. EMBO Mol. Med. 2021, 13, e14365. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Uppal, A.; Yusuf, J.; Muheeb, G.; Agarwal, R. COVID-19 induced ventricular tachycardia storm unmasking a clinically silent cardiomyopathy: A case report. Eur. Heart J. Case Rep. 2021, 5, ytab220. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasireddi, S.K.; Sattayaprasert, P.; Yang, D.; Dennis, A.T.; Bektik, E.; Fu, J.-d.; Mackall, J.A.; Laurita, K.R. Adipogenic Signaling Promotes Arrhythmia Substrates before Structural Abnormalities in TMEM43 ARVC. J. Pers. Med. 2022, 12, 1680. https://doi.org/10.3390/jpm12101680
Vasireddi SK, Sattayaprasert P, Yang D, Dennis AT, Bektik E, Fu J-d, Mackall JA, Laurita KR. Adipogenic Signaling Promotes Arrhythmia Substrates before Structural Abnormalities in TMEM43 ARVC. Journal of Personalized Medicine. 2022; 12(10):1680. https://doi.org/10.3390/jpm12101680
Chicago/Turabian StyleVasireddi, Sunil K., Prasongchai Sattayaprasert, Dandan Yang, Adrienne T. Dennis, Emre Bektik, Ji-dong Fu, Judith A. Mackall, and Kenneth R. Laurita. 2022. "Adipogenic Signaling Promotes Arrhythmia Substrates before Structural Abnormalities in TMEM43 ARVC" Journal of Personalized Medicine 12, no. 10: 1680. https://doi.org/10.3390/jpm12101680
APA StyleVasireddi, S. K., Sattayaprasert, P., Yang, D., Dennis, A. T., Bektik, E., Fu, J.-d., Mackall, J. A., & Laurita, K. R. (2022). Adipogenic Signaling Promotes Arrhythmia Substrates before Structural Abnormalities in TMEM43 ARVC. Journal of Personalized Medicine, 12(10), 1680. https://doi.org/10.3390/jpm12101680