
Short-Chain Fatty Acids in Gut–Heart Axis: Their Role in the Pathology of Heart Failure


 ,
,
Abstract
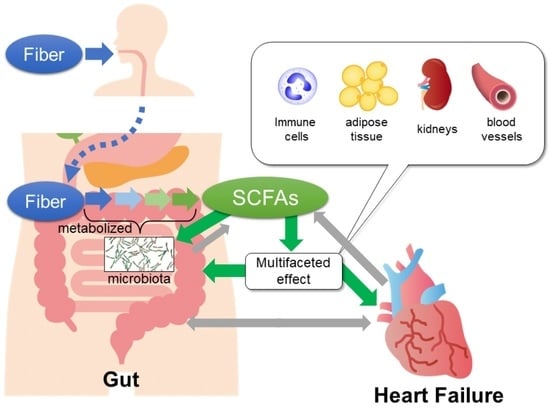
{kind=link}
{kind=link}
1. Introduction
2. The Molecular Action of SCFA
2.1. HDAC Inhibition by SCFA
2.2. SCFAs’ Effect via G-Protein-Coupled Receptors (GPRs)
3. The Change of SCFA and Related Signals in Cardiovascular Diseases including HF
4. The Potential Mechanisms of SCFAs’ Action in the Pathology of HF
4.1. SCFAs Restore Mitochondrial Function
4.2. SCFAs Regulate Blood Pressure
4.3. SCFA’s Effect on Cardiac Inflammatory Response
4.4. SCFA as a Fuel Source
4.5. SCFAs’ Effect on Gut
4.6. SCFAs’ Effect on Adipose Tissue and Adipokines
4.7. SCFAs’ Effect on Kidneys
4.8. Negative Effect of SCFAs
5. Clinical Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Nagatomo, Y.; Tang, W.H. Intersections Between Microbiome and Heart Failure: Revisiting the Gut Hypothesis. J. Card. Fail. 2015, 21, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Alverdy, J.; Zaborina, O.; Wu, L. The impact of stress and nutrition on bacterial-host interactions at the intestinal epithelial surface. Curr. Opin. Clin. Nutr. Metab. Care 2005, 8, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, Q.; He, Q.; Geng, Y.; Tang, C.; Wang, C.; Li, J. Temporal variations of the ileal microbiota in intestinal ischemia and reperfusion. Shock 2013, 39, 96–103. [Google Scholar] [CrossRef]
- Wang, F.; Li, Q.; Wang, C.; Tang, C.; Li, J. Dynamic alteration of the colonic microbiota in intestinal ischemia-reperfusion injury. PLoS ONE 2012, 7, e42027. [Google Scholar] [CrossRef]
- Llamas, M.A.; Aller, M.A.; Marquina, D.; Nava, M.P.; Arias, J. Bacterial translocation to mesenteric lymph nodes increases in chronic portal hypertensive rats. Dig. Dis. Sci. 2010, 55, 2244–2254. [Google Scholar] [CrossRef]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef]
- Roy, C.C.; Kien, C.L.; Bouthillier, L.; Levy, E. Short-chain fatty acids: Ready for prime time? Nutr. Clin. Pract. 2006, 21, 351–366. [Google Scholar] [CrossRef]
- Høverstad, T.; Midtvedt, T. Short-chain fatty acids in germfree mice and rats. J. Nutr. 1986, 116, 1772–1776. [Google Scholar] [CrossRef]
- Tazoe, H.; Otomo, Y.; Kaji, I.; Tanaka, R.; Karaki, S.I.; Kuwahara, A. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J. Physiol. Pharmacol. 2008, 59 (Suppl. S2), 251–262. [Google Scholar]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Weaver, G.A.; Krause, J.A.; Miller, T.L.; Wolin, M.J. Short chain fatty acid distributions of enema samples from a sigmoidoscopy population: An association of high acetate and low butyrate ratios with adenomatous polyps and colon cancer. Gut 1988, 29, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef]
- Lu, J.; McKinsey, T.A.; Nicol, R.L.; Olson, E.N. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 4070–4075. [Google Scholar] [CrossRef]
- Gusterson, R.J.; Jazrawi, E.; Adcock, I.M.; Latchman, D.S. The transcriptional co-activators CREB-binding protein (CBP) and p300 play a critical role in cardiac hypertrophy that is dependent on their histone acetyltransferase activity. J. Biol. Chem. 2003, 278, 6838–6847. [Google Scholar] [CrossRef]
- Antos, C.L.; McKinsey, T.A.; Dreitz, M.; Hollingsworth, L.M.; Zhang, C.L.; Schreiber, K.; Rindt, H.; Gorczynski, R.J.; Olson, E.N. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J. Biol. Chem. 2003, 278, 28930–28937. [Google Scholar] [CrossRef]
- Ooi, J.Y.; Tuano, N.K.; Rafehi, H.; Gao, X.M.; Ziemann, M.; Du, X.J.; El-Osta, A. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics 2015, 10, 418–430. [Google Scholar] [CrossRef]
- Kong, Y.; Tannous, P.; Lu, G.; Berenji, K.; Rothermel, B.A.; Olson, E.N.; Hill, J.A. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 2006, 113, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Guan, X.; Wang, X.; Qu, S.; Zhang, S.; Hui, W.; Men, L.; Chen, X. Dynamic modulation of gut microbiota improves post-myocardial infarct tissue repair in rats via butyric acid-mediated histone deacetylase inhibition. FASEB J. 2021, 35, e21385. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Seok, Y.M.; Song, M.J.; Lee, H.A.; Kurz, T.; Kim, I. Histone deacetylase inhibition attenuates cardiac hypertrophy and fibrosis through acetylation of mineralocorticoid receptor in spontaneously hypertensive rats. Mol. Pharmacol. 2015, 87, 782–791. [Google Scholar] [CrossRef]
- Xie, M.; Kong, Y.; Tan, W.; May, H.; Battiprolu, P.K.; Pedrozo, Z.; Wang, Z.V.; Morales, C.; Luo, X.; Cho, G.; et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 2014, 129, 1139–1151. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.J.; Loh, Y.W.; Singer, J.; Zhu, W.; Macia, L.; Mackay, C.R.; Wang, W.; Chadban, S.J.; Wu, H. Fiber Derived Microbial Metabolites Prevent Acute Kidney Injury Through G-Protein Coupled Receptors and HDAC Inhibition. Front. Cell Dev. Biol. 2021, 9, 648639. [Google Scholar] [CrossRef]
- Vinolo, M.A.; Rodrigues, H.G.; Hatanaka, E.; Sato, F.T.; Sampaio, S.C.; Curi, R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J. Nutr. Biochem. 2011, 22, 849–855. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The role of short-chain fatty acids in health and disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar] [CrossRef]
- Usami, M.; Kishimoto, K.; Ohata, A.; Miyoshi, M.; Aoyama, M.; Fueda, Y.; Kotani, J. Butyrate and trichostatin A attenuate nuclear factor kappaB activation and tumor necrosis factor alpha secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr. Res. 2008, 28, 321–328. [Google Scholar] [CrossRef]
- Kasubuchi, M.; Hasegawa, S.; Hiramatsu, T.; Ichimura, A.; Kimura, I. Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients 2015, 7, 2839–2849. [Google Scholar] [CrossRef]
- Miyamoto, J.; Hasegawa, S.; Kasubuchi, M.; Ichimura, A.; Nakajima, A.; Kimura, I. Nutritional Signaling via Free Fatty Acid Receptors. Int. J. Mol. Sci. 2016, 17, 450. [Google Scholar] [CrossRef] [PubMed]
- Tazoe, H.; Otomo, Y.; Karaki, S.; Kato, I.; Fukami, Y.; Terasaki, M.; Kuwahara, A. Expression of short-chain fatty acid receptor GPR41 in the human colon. Biomed. Res. 2009, 30, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Tunaru, S.; Kero, J.; Schaub, A.; Wufka, C.; Blaukat, A.; Pfeffer, K.; Offermanns, S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat. Med. 2003, 9, 352–355. [Google Scholar] [CrossRef]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- Spehr, M.; Gisselmann, G.; Poplawski, A.; Riffell, J.A.; Wetzel, C.H.; Zimmer, R.K.; Hatt, H. Identification of a testicular odorant receptor mediating human sperm chemotaxis. Science 2003, 299, 2054–2058. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Suster, M.S.; Borges, J.I. Short-Chain Fatty Acid Receptors and Cardiovascular Function. Int. J. Mol. Sci. 2022, 23, 3303. [Google Scholar] [CrossRef]
- Kaye, D.M.; Shihata, W.A.; Jama, H.A.; Tsyganov, K.; Ziemann, M.; Kiriazis, H.; Horlock, D.; Vijay, A.; Giam, B.; Vinh, A.; et al. Deficiency of Prebiotic Fiber and Insufficient Signaling Through Gut Metabolite-Sensing Receptors Leads to Cardiovascular Disease. Circulation 2020, 141, 1393–1403. [Google Scholar] [CrossRef]
- Natarajan, N.; Hori, D.; Flavahan, S.; Steppan, J.; Flavahan, N.A.; Berkowitz, D.E.; Pluznick, J.L. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol. Genom. 2016, 48, 826–834. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.K.; McKenzie, C.; Mariño, E.; Macia, L.; Mackay, C.R. Metabolite-Sensing G Protein-Coupled Receptors-Facilitators of Diet-Related Immune Regulation. Annu. Rev. Immunol. 2017, 35, 371–402. [Google Scholar] [CrossRef]
- Kim, S.; Goel, R.; Kumar, A.; Qi, Y.; Lobaton, G.; Hosaka, K.; Mohammed, M.; Handberg, E.M.; Richards, E.M.; Pepine, C.J.; et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. 2018, 132, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhou, C.; Chen, Y.; He, X.; Gao, F.; Xue, D. Quantitative increase in short-chain fatty acids, especially butyrate protects kidney from ischemia/reperfusion injury. J. Investig. Med. 2022, 70, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Zuo, K.; Fang, C.; Liu, Z.; Fu, Y.; Liu, Y.; Liu, L.; Wang, Y.; Yin, X.; Liu, X.; Li, J.; et al. Commensal microbe-derived SCFA alleviates atrial fibrillation via GPR43/NLRP3 signaling. Int. J. Biol. Sci. 2022, 18, 4219–4232. [Google Scholar] [CrossRef]
- Kamo, T.; Akazawa, H.; Suda, W.; Saga-Kamo, A.; Shimizu, Y.; Yagi, H.; Liu, Q.; Nomura, S.; Naito, A.T.; Takeda, N.; et al. Dysbiosis and compositional alterations with aging in the gut microbiota of patients with heart failure. PLoS ONE 2017, 12, e0174099. [Google Scholar] [CrossRef]
- Luedde, M.; Winkler, T.; Heinsen, F.A.; Rühlemann, M.C.; Spehlmann, M.E.; Bajrovic, A.; Lieb, W.; Franke, A.; Ott, S.J.; Frey, N. Heart failure is associated with depletion of core intestinal microbiota. ESC Heart Fail. 2017, 4, 282–290. [Google Scholar] [CrossRef]
- Endo, H.; Dohi, T.; Funamizu, T.; Shitara, J.; Wada, H.; Doi, S.; Naito, R.; Konishi, H.; Ogita, M.; Iwata, H.; et al. Long-Term Predictive Value of High-Sensitivity C-Reactive Protein for Cancer Mortality in Patients Undergoing Percutaneous Coronary Intervention. Circ. J. 2019, 83, 630–636. [Google Scholar] [CrossRef]
- Yan, Q.; Gu, Y.; Li, X.; Yang, W.; Jia, L.; Chen, C.; Han, X.; Huang, Y.; Zhao, L.; Li, P.; et al. Alterations of the Gut Microbiome in Hypertension. Front. Cell. Infect. Microbiol. 2017, 7, 381. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef]
- Li, X.; Li, R.; You, N.; Zhao, X.; Li, J.; Jiang, W. Butyric Acid Ameliorates Myocardial Fibrosis by Regulating M1/M2 Polarization of Macrophages and Promoting Recovery of Mitochondrial Function. Front. Nutr. 2022, 9, 875473. [Google Scholar] [CrossRef] [PubMed]
- Nakai, M.; Ribeiro, R.V.; Stevens, B.R.; Gill, P.; Muralitharan, R.R.; Yiallourou, S.; Muir, J.; Carrington, M.; Head, G.A.; Kaye, D.M.; et al. Essential Hypertension Is Associated With Changes in Gut Microbial Metabolic Pathways: A Multisite Analysis of Ambulatory Blood Pressure. Hypertension 2021, 78, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hua, Y.; Ren, J. Short-chain fatty acid propionate alleviates Akt2 knockout-induced myocardial contractile dysfunction. Exp. Diabetes Res. 2012, 2012, 851717. [Google Scholar] [CrossRef] [PubMed]
- Yuzefpolskaya, M.; Bohn, B.; Javaid, A.; Mondellini, G.M.; Braghieri, L.; Pinsino, A.; Onat, D.; Cagliostro, B.; Kim, A.; Takeda, K.; et al. Levels of Trimethylamine N-Oxide Remain Elevated Long Term After Left Ventricular Assist Device and Heart Transplantation and Are Independent From Measures of Inflammation and Gut Dysbiosis. Circ. Heart Fail. 2021, 14, e007909. [Google Scholar] [CrossRef] [PubMed]
- Yuzefpolskaya, M.; Bohn, B.; Nasiri, M.; Zuver, A.M.; Onat, D.D.; Royzman, E.A.; Nwokocha, J.; Mabasa, M.; Pinsino, A.; Brunjes, D.; et al. Gut microbiota, endotoxemia, inflammation, and oxidative stress in patients with heart failure, left ventricular assist device, and transplant. J. Heart Lung Transplant. 2020, 39, 880–890. [Google Scholar] [CrossRef]
- Kessler-Icekson, G.; Hochhauser, E.; Sinai, T.; Kremer, A.; Dick, J.; Tarasenko, N.; Nudelman, V.; Schlesinger, H.; Abraham, S.; Nudelman, A.; et al. A histone deacetylase inhibitory prodrug-butyroyloxymethyl diethyl phosphate-protects the heart and cardiomyocytes against ischemia injury. Eur. J. Pharm. Sci. 2012, 45, 592–599. [Google Scholar] [CrossRef]
- Nudelman, V.; Zahalka, M.A.; Nudelman, A.; Rephaeli, A.; Kessler-Icekson, G. Cardioprotection by AN-7, a prodrug of the histone deacetylase inhibitor butyric acid: Selective activity in hypoxic cardiomyocytes and cardiofibroblasts. Eur. J. Pharmacol. 2020, 882, 173255. [Google Scholar] [CrossRef]
- Rephaeli, A.; Waks-Yona, S.; Nudelman, A.; Tarasenko, I.; Tarasenko, N.; Phillips, D.R.; Cutts, S.M.; Kessler-Icekson, G. Anticancer prodrugs of butyric acid and formaldehyde protect against doxorubicin-induced cardiotoxicity. Br. J. Cancer 2007, 96, 1667–1674. [Google Scholar] [CrossRef]
- Russo, M.; Guida, F.; Paparo, L.; Trinchese, G.; Aitoro, R.; Avagliano, C.; Fiordelisi, A.; Napolitano, F.; Mercurio, V.; Sala, V.; et al. The novel butyrate derivative phenylalanine-butyramide protects from doxorubicin-induced cardiotoxicity. Eur. J. Heart Fail. 2019, 21, 519–528. [Google Scholar] [CrossRef]
- Huang, J.; Wei, S.; Jiang, C.; Xiao, Z.; Liu, J.; Peng, W.; Zhang, B.; Li, W. Involvement of Abnormal Gut Microbiota Composition and Function in Doxorubicin-Induced Cardiotoxicity. Front. Cell. Infect. Microbiol. 2022, 12, 808837. [Google Scholar] [CrossRef]
- Sharman, J.E.; O′Brien, E.; Alpert, B.; Schutte, A.E.; Delles, C.; Hecht Olsen, M.; Asmar, R.; Atkins, N.; Barbosa, E.; Calhoun, D.; et al. Lancet Commission on Hypertension group position statement on the global improvement of accuracy standards for devices that measure blood pressure. J. Hypertens. 2020, 38, 21–29. [Google Scholar] [CrossRef]
- Alonso, A.; de la Fuente, C.; Martín-Arnau, A.M.; de Irala, J.; Martínez, J.A.; Martínez-González, M.A. Fruit and vegetable consumption is inversely associated with blood pressure in a Mediterranean population with a high vegetable-fat intake: The Seguimiento Universidad de Navarra (SUN) Study. Br. J. Nutr. 2004, 92, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Whelton, S.P.; Hyre, A.D.; Pedersen, B.; Yi, Y.; Whelton, P.K.; He, J. Effect of dietary fiber intake on blood pressure: A meta-analysis of randomized, controlled clinical trials. J. Hypertens. 2005, 23, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Streppel, M.T.; Arends, L.R.; van′t Veer, P.; Grobbee, D.E.; Geleijnse, J.M. Dietary fiber and blood pressure: A meta-analysis of randomized placebo-controlled trials. Arch. Intern. Med. 2005, 165, 150–156. [Google Scholar] [CrossRef]
- Daugirdas, J.T.; Nawab, Z.M. Acetate relaxation of isolated vascular smooth muscle. Kidney Int. 1987, 32, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Nutting, C.W.; Islam, S.; Daugirdas, J.T. Vasorelaxant effects of short chain fatty acid salts in rat caudal artery. Am. J. Physiol. Circ. Physiol. 1991, 261, H561–H567. [Google Scholar] [CrossRef]
- Mortensen, F.V.; Nielsen, H.; Mulvany, M.J.; Hessov, I. Short chain fatty acids dilate isolated human colonic resistance arteries. Gut 1990, 31, 1391–1394. [Google Scholar] [CrossRef]
- Pluznick, J.L.; Protzko, R.J.; Gevorgyan, H.; Peterlin, Z.; Sipos, A.; Han, J.; Brunet, I.; Wan, L.X.; Rey, F.; Wang, T.; et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 4410–4415. [Google Scholar] [CrossRef]
- Bartolomaeus, H.; Balogh, A.; Yakoub, M.; Homann, S.; Markó, L.; Höges, S.; Tsvetkov, D.; Krannich, A.; Wundersitz, S.; Avery, E.G.; et al. Short-Chain Fatty Acid Propionate Protects From Hypertensive Cardiovascular Damage. Circulation 2019, 139, 1407–1421. [Google Scholar] [CrossRef]
- Palm, C.L.; Nijholt, K.T.; Bakker, B.M.; Westenbrink, B.D. Short-Chain Fatty Acids in the Metabolism of Heart Failure-Rethinking the Fat Stigma. Front. Cardiovasc. Med. 2022, 9, 915102. [Google Scholar] [CrossRef]
- Lewandowski, E.D.; Kudej, R.K.; White, L.T.; O’Donnell, J.M.; Vatner, S.F. Mitochondrial preference for short chain fatty acid oxidation during coronary artery constriction. Circulation 2002, 105, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Carley, A.N.; Maurya, S.K.; Fasano, M.; Wang, Y.; Selzman, C.H.; Drakos, S.G.; Lewandowski, E.D. Short-Chain Fatty Acids Outpace Ketone Oxidation in the Failing Heart. Circulation 2021, 143, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Sandek, A.; Bauditz, J.; Swidsinski, A.; Buhner, S.; Weber-Eibel, J.; von Haehling, S.; Schroedl, W.; Karhausen, T.; Doehner, W.; Rauchhaus, M.; et al. Altered intestinal function in patients with chronic heart failure. J. Am. Coll. Cardiol. 2007, 50, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.A.; Srinivas, M.; Shariff, A.; Nazir, T. Role of short chain fatty acids in mesenteric ischemia reperfusion injury in rats. Eur. J. Pediatr. Surg. 2010, 20, 98–101. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Yan, H.; Ajuwon, K.M. Butyrate modifies intestinal barrier function in IPEC-J2 cells through a selective upregulation of tight junction proteins and activation of the Akt signaling pathway. PLoS ONE 2017, 12, e0179586. [Google Scholar] [CrossRef]
- Ohata, A.; Usami, M.; Miyoshi, M. Short-chain fatty acids alter tight junction permeability in intestinal monolayer cells via lipoxygenase activation. Nutrition 2005, 21, 838–847. [Google Scholar] [CrossRef]
- Ferreira, T.M.; Leonel, A.J.; Melo, M.A.; Santos, R.R.; Cara, D.C.; Cardoso, V.N.; Correia, M.I.; Alvarez-Leite, J.I. Oral supplementation of butyrate reduces mucositis and intestinal permeability associated with 5-Fluorouracil administration. Lipids 2012, 47, 669–678. [Google Scholar] [CrossRef]
- Kibbie, J.J.; Dillon, S.M.; Thompson, T.A.; Purba, C.M.; McCarter, M.D.; Wilson, C.C. Butyrate directly decreases human gut lamina propria CD4 T cell function through histone deacetylase (HDAC) inhibition and GPR43 signaling. Immunobiology 2021, 226, 152126. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Jama, H.A.; Fiedler, A.; Tsyganov, K.; Nelson, E.; Horlock, D.; Nakai, M.E.; Kiriazis, H.; Johnson, C.; Du, X.J.; Mackay, C.R.; et al. Manipulation of the gut microbiota by the use of prebiotic fibre does not override a genetic predisposition to heart failure. Sci. Rep. 2020, 10, 17919. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, H.; Obokata, M.; Omote, K.; Reddy, Y.N.V.; Takahashi, N.; Koepp, K.E.; Ng, A.C.T.; Rider, O.J.; Borlaug, B.A. Long-Term Changes in Cardiac Structure and Function Following Bariatric Surgery. J. Am. Coll. Cardiol. 2022, 80, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Feraco, A.; Marzolla, V.; Scuteri, A.; Armani, A.; Caprio, M. Mineralocorticoid Receptors in Metabolic Syndrome: From Physiology to Disease. Trends Endocrinol. Metab. 2020, 31, 205–217. [Google Scholar] [CrossRef]
- Faulkner, J.L.; Bruder-Nascimento, T.; Belin de Chantemèle, E.J. The regulation of aldosterone secretion by leptin: Implications in obesity-related cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 2018, 27, 63–69. [Google Scholar] [CrossRef]
- Loffreda, S.; Yang, S.Q.; Lin, H.Z.; Karp, C.L.; Brengman, M.L.; Wang, D.J.; Klein, A.S.; Bulkley, G.B.; Bao, C.; Noble, P.W.; et al. Leptin regulates proinflammatory immune responses. FASEB J. 1998, 12, 57–65. [Google Scholar] [CrossRef]
- Vatutin, N.T.; Shevelok, A.N. Relationship between Blood Aldosterone and Somatometric Parameters in Patients with Chronic Heart Failure and Preserved Ejection Fraction of Left Ventricle. Klin Med. (Mosk) 2016, 94, 265–269. [Google Scholar] [CrossRef]
- Van den Bergh, A.; Vanderper, A.; Vangheluwe, P.; Desjardins, F.; Nevelsteen, I.; Verreth, W.; Wuytack, F.; Holvoet, P.; Flameng, W.; Balligand, J.L.; et al. Dyslipidaemia in type II diabetic mice does not aggravate contractile impairment but increases ventricular stiffness. Cardiovasc. Res. 2008, 77, 371–379. [Google Scholar] [CrossRef]
- Ouchi, N.; Kihara, S.; Arita, Y.; Maeda, K.; Kuriyama, H.; Okamoto, Y.; Hotta, K.; Nishida, M.; Takahashi, M.; Nakamura, T.; et al. Novel modulator for endothelial adhesion molecules: Adipocyte-derived plasma protein adiponectin. Circulation 1999, 100, 2473–2476. [Google Scholar] [CrossRef]
- Antuna-Puente, B.; Feve, B.; Fellahi, S.; Bastard, J.P. Adipokines: The missing link between insulin resistance and obesity. Diabetes Metab. 2008, 34, 2–11. [Google Scholar] [CrossRef]
- Essick, E.E.; Wilson, R.M.; Pimentel, D.R.; Shimano, M.; Baid, S.; Ouchi, N.; Sam, F. Adiponectin modulates oxidative stress-induced autophagy in cardiomyocytes. PLoS ONE 2013, 8, e68697. [Google Scholar] [CrossRef] [PubMed]
- Essick, E.E.; Ouchi, N.; Wilson, R.M.; Ohashi, K.; Ghobrial, J.; Shibata, R.; Pimentel, D.R.; Sam, F. Adiponectin mediates cardioprotection in oxidative stress-induced cardiac myocyte remodeling. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H984–H993. [Google Scholar] [CrossRef]
- Tesauro, M.; Canale, M.P.; Rodia, G.; Di Daniele, N.; Lauro, D.; Scuteri, A.; Cardillo, C. Metabolic syndrome, chronic kidney, and cardiovascular diseases: Role of adipokines. Cardiol. Res. Pract. 2011, 2011, 653182. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arter. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.J.; Park, C.G.; Seo, H.S.; Oh, D.J.; Ro, Y.M. Associations among plasma adiponectin, hypertension, left ventricular diastolic function and left ventricular mass index. Blood Press. 2004, 13, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Kronenberg, F.; Kielstein, J.T.; Haller, H.; Morath, C.; Ritz, E.; Fliser, D. Renal insulin resistance syndrome, adiponectin and cardiovascular events in patients with kidney disease: The mild and moderate kidney disease study. J. Am. Soc. Nephrol. 2005, 16, 1091–1098. [Google Scholar] [CrossRef]
- Pelgrim, C.E.; Franx, B.A.A.; Snabel, J.; Kleemann, R.; Arnoldussen, I.A.C.; Kiliaan, A.J. Butyrate Reduces HFD-Induced Adipocyte Hypertrophy and Metabolic Risk Factors in Obese LDLr-/-.Leiden Mice. Nutrients 2017, 9, 714. [Google Scholar] [CrossRef]
- Lu, Y.; Fan, C.; Liang, A.; Fan, X.; Wang, R.; Li, P.; Qi, K. Effects of SCFA on the DNA methylation pattern of adiponectin and resistin in high-fat-diet-induced obese male mice. Br. J. Nutr. 2018, 120, 385–392. [Google Scholar] [CrossRef]
- Lamacchia, O.; Nicastro, V.; Camarchio, D.; Valente, U.; Grisorio, R.; Gesualdo, L.; Cignarelli, M. Para- and perirenal fat thickness is an independent predictor of chronic kidney disease, increased renal resistance index and hyperuricaemia in type-2 diabetic patients. Nephrol. Dial. Transplant. 2011, 26, 892–898. [Google Scholar] [CrossRef]
- Papafragkaki, D.K.; Tolis, G. Obesity and renal disease: A possible role of leptin. Hormones 2005, 4, 90–95. [Google Scholar]
- Gonzalez, A.; Krieg, R.; Massey, H.D.; Carl, D.; Ghosh, S.; Gehr, T.W.B.; Ghosh, S.S. Sodium butyrate ameliorates insulin resistance and renal failure in CKD rats by modulating intestinal permeability and mucin expression. Nephrol. Dial. Transplant. 2019, 34, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Yu, H.; Chen, J.; Mok, S.W.F.; Tan, X.; Zhao, B.; He, S.; Lan, L.; Fu, X.; Chen, G.; et al. The short-chain fatty acid butyrate accelerates vascular calcification via regulation of histone deacetylases and NF-κB signaling. Vasc. Pharmacol. 2022, 146, 107096. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yukino-Iwashita, M.; Nagatomo, Y.; Kawai, A.; Taruoka, A.; Yumita, Y.; Kagami, K.; Yasuda, R.; Toya, T.; Ikegami, Y.; Masaki, N.; et al. Short-Chain Fatty Acids in Gut–Heart Axis: Their Role in the Pathology of Heart Failure. J. Pers. Med. 2022, 12, 1805. https://doi.org/10.3390/jpm12111805
Yukino-Iwashita M, Nagatomo Y, Kawai A, Taruoka A, Yumita Y, Kagami K, Yasuda R, Toya T, Ikegami Y, Masaki N, et al. Short-Chain Fatty Acids in Gut–Heart Axis: Their Role in the Pathology of Heart Failure. Journal of Personalized Medicine. 2022; 12(11):1805. https://doi.org/10.3390/jpm12111805
Chicago/Turabian StyleYukino-Iwashita, Midori, Yuji Nagatomo, Akane Kawai, Akira Taruoka, Yusuke Yumita, Kazuki Kagami, Risako Yasuda, Takumi Toya, Yukinori Ikegami, Nobuyuki Masaki, and et al. 2022. "Short-Chain Fatty Acids in Gut–Heart Axis: Their Role in the Pathology of Heart Failure" Journal of Personalized Medicine 12, no. 11: 1805. https://doi.org/10.3390/jpm12111805
APA StyleYukino-Iwashita, M., Nagatomo, Y., Kawai, A., Taruoka, A., Yumita, Y., Kagami, K., Yasuda, R., Toya, T., Ikegami, Y., Masaki, N., Ido, Y., & Adachi, T. (2022). Short-Chain Fatty Acids in Gut–Heart Axis: Their Role in the Pathology of Heart Failure. Journal of Personalized Medicine, 12(11), 1805. https://doi.org/10.3390/jpm12111805