
Systematic Analysis of Cellular Signaling Pathways and Therapeutic Targets for SLC45A3:ERG Fusion-Positive Prostate Cancer


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Data Acquisition
2.2. Case and Control Sample Selection
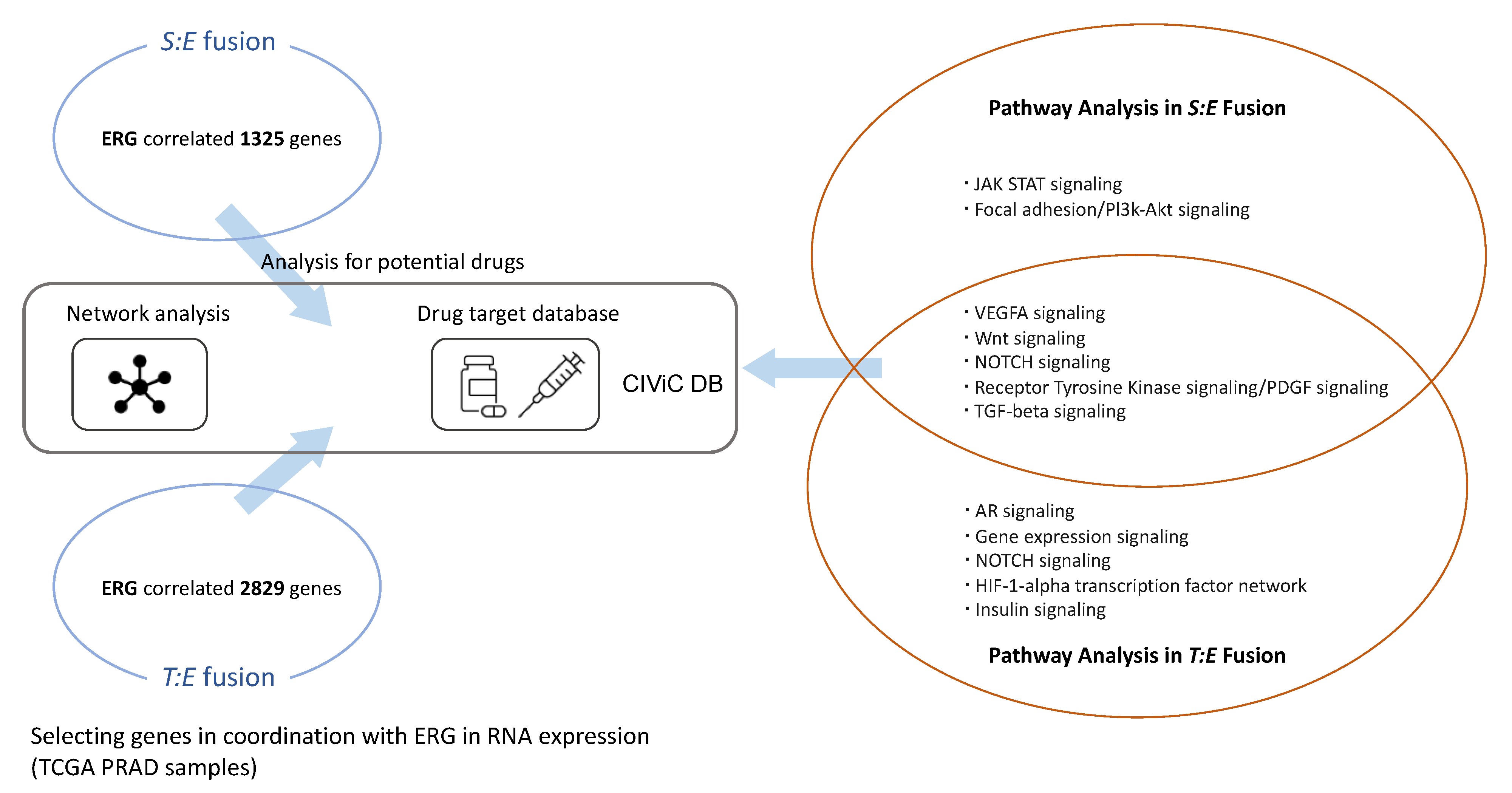
2.3. Selection of Genes Related to S:E and T:E Fusion
2.4. Pathway Analysis via ConsensusPathDB (CPDB) and Over-Representation
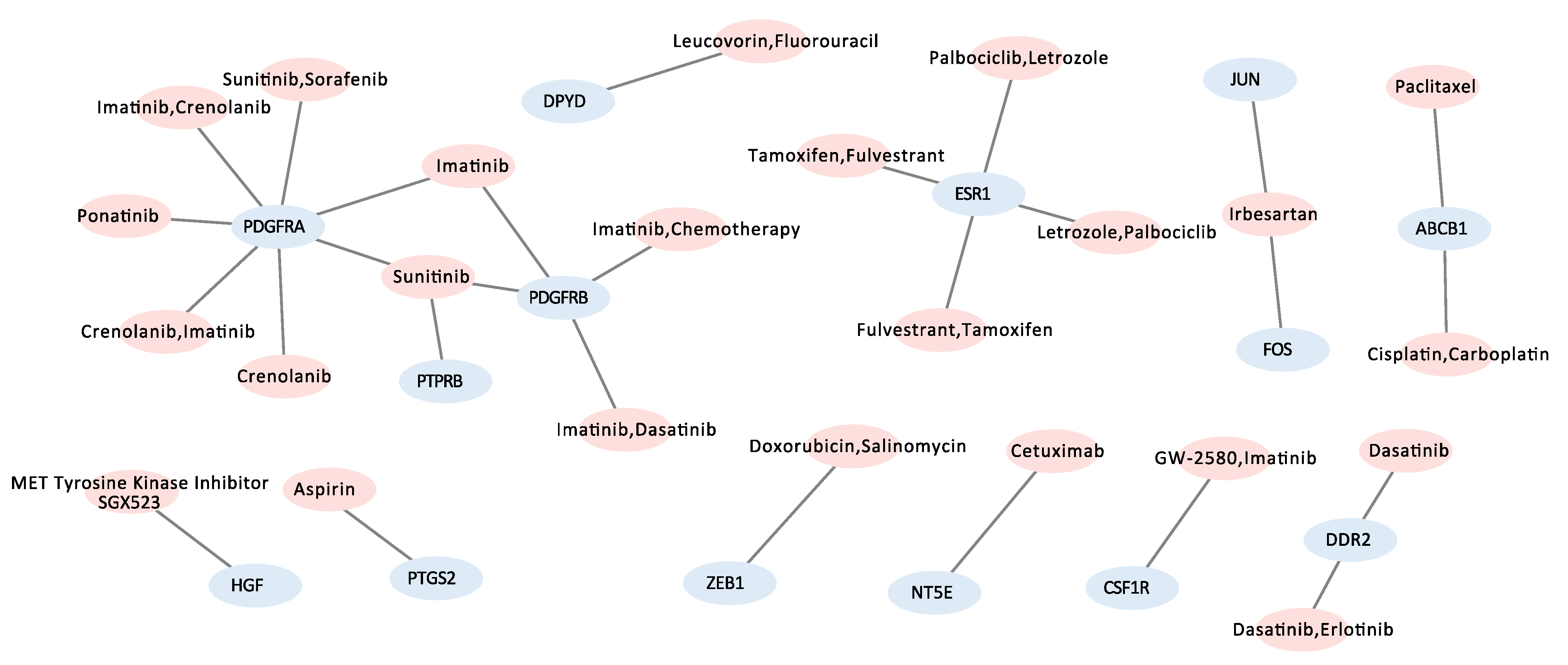
2.5. Organization of Gene-Drug Network via CIViC
2.6. Data Visualization and Analysis via R
3. Results
3.1. Clinical Characteristics of Prostate Cancer Patients
3.2. Cellular Pathways Associated with S:E Fusion-Positive Prostate Cancer
3.3. Cellular Pathways Associated with T:E Fusion-Positive Prostate Cancer
3.4. Comparison of Cellular Pathways Involved in S:E Fusion-Positive Prostate Cancer vs. T:E Fusion-Positive Prostate Cancer
3.5. Candidate Target Genes and Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Adamo, P.; Ladomery, M.R. The oncogene ERG: A key factor in prostate cancer. Oncogene 2016, 35, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Bjartell, A.; Chinnaiyan, A.M.; Jenster, G.; Nam, R.K.; Rubin, M.; Schalken, J.A. ETS Gene Fusions in Prostate Cancer: From Discovery to Daily Clinical Practice. Eur. Urol. 2009, 56, 275–286. [Google Scholar] [CrossRef]
- Hong, Z.; Zhang, W.; Ding, D.; Huang, Z.; Yan, Y.; Cao, W.; Pan, Y.; Hou, X.; Weroha, S.J.; Karnes, R.J.; et al. DNA Damage Promotes TMPRSS2-ERG Oncoprotein Destruction and Prostate Cancer Suppression via Signaling Converged by GSK3β and WEE1. Mol. Cell 2020, 79, 1008–1023.e4. [Google Scholar] [CrossRef]
- Gordanpour, A.; Stanimirovic, A.; Nam, R.K.; Moreno, C.S.; Sherman, C.; Sugar, L.; Seth, A. miR-221 Is down-regulated in TMPRSS2:ERG fusion-positive prostate cancer. Anticancer Res. 2011, 31, 403–410. [Google Scholar] [PubMed]
- Mohamed, A.A.; Xavier, C.P.; Sukumar, G.; Tan, S.-H.; Ravindranath, L.; Seraj, N.; Kumar, V.; Sreenath, T.L.; McLeod, D.G.; Petrovics, G.; et al. Identification of a Small Molecule That Selectively Inhibits ERG-Positive Cancer Cell Growth. Cancer Res. 2018, 78, 3659–3671. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.W.; Lee, S.; Chun, S.; Lee, K.W.; Kim, J.; Kim, H.S. Comprehensive analysis of oncogenic signatures and consequent repurposed drugs in TMPRSS2:ERG fusion-positive prostate cancer. Clin. Transl. Med. 2021, 11, e420. [Google Scholar] [CrossRef]
- Esgueva, R.; Perner, S.; LaFargue, C.J.; Scheble, V.; Stephan, C.; Lein, M.; Fritzsche, F.R.; Dietel, M.; Kristiansen, G.; Rubin, M.A. Prevalence of TMPRSS2–ERG and SLC45A3–ERG gene fusions in a large prostatectomy cohort. Mod. Pathol. 2010, 23, 539–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamburov, A.; Herwig, R. ConsensusPathDB 2022: Molecular interactions update as a resource for network biology. Nucleic Acids Res. 2022, 50, D587–D595. [Google Scholar] [CrossRef]
- Clark, J.; Merson, S.; Jhavar, S.; Flohr, P.; Edwards, S.; Foster, C.S.; Eeles, R.; Martin, F.L.; Phillips, D.H.; Crundwell, M.; et al. Diversity of TMPRSS2-ERG fusion transcripts in the human prostate. Oncogene 2007, 26, 2667–2673. [Google Scholar] [CrossRef]
- Murillo-Garzón, V.; Gorroño-Etxebarria, I.; Åkerfelt, M.; Puustinen, M.C.; Sistonen, L.; Nees, M.; Carton, J.; Waxman, J.; Kypta, R.M. Frizzled-8 integrates Wnt-11 and transforming growth factor-β signaling in prostate cancer. Nat. Commun. 2018, 9, 1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarthi, B.V.S.K.; Chandrashekar, D.S.; Balasubramanya, S.A.H.; Robinson, A.; Carskadon, S.; Rao, U.; Gordetsky, J.; Manne, U.; Netto, G.J.; Sudarshan, S.; et al. Wnt receptor Frizzled 8 is a target of ERG in prostate cancer. Prostate 2018, 78, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Loizzo, D.; Pandolfo, S.D.; Rogers, D.; Cerrato, C.; di Meo, N.A.; Autorino, R.; Mirone, V.; Ferro, M.; Porta, C.; Stella, A.; et al. Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 3826. [Google Scholar] [CrossRef]
- Menges, C.W.; Altomare, D.A.; Testa, J.R. FAS-associated factor 1 (FAF1): Diverse functions and implications for oncogenesis. Cell Cycle 2009, 8, 2528–2534. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, M.C.; Deryugina, E.I.; Suarez, E.; Lopez, S.M.; Lin, D.; Xue, H.; Gorlov, I.P.; Wang, Y.; Agoulnik, I.U. INPP4B suppresses prostate cancer cell invasion. Cell Commun. Signal. 2014, 12, 61. [Google Scholar] [CrossRef]
- Saitoh, T.; Moriwaki, J.; Koike, J.; Takagi, A.; Miwa, T.; Shiokawa, K.; Katoh, M. Molecular Cloning and Characterization of FRAT2, Encoding a Positive Regulator of the WNT Signaling Pathway. Biochem. Biophys. Res. Commun. 2001, 281, 815–820. [Google Scholar] [CrossRef]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.G.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.D.; Glass, C.K.; et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef]
- Watanabe, R.; Maekawa, M.; Hieda, M.; Taguchi, T.; Miura, N.; Kikugawa, T.; Saika, T.; Higashiyama, S. SPOP is essential for DNA–protein cross-link repair in prostate cancer cells: SPOP-dependent removal of topoisomerase 2A from the topoisomerase 2A-DNA cleavage complex. Mol. Biol. Cell 2020, 31, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Parolia, A.; Cieslik, M.; Chu, S.-C.; Xiao, L.; Ouchi, T.; Zhang, Y.; Wang, X.; Vats, P.; Cao, X.; Pitchiaya, S.; et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 2019, 571, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhu, Y.; Ma, J.; Chang, K.; Ding, D.; Bai, Y.; Gao, K.; Zhang, P.; Mo, R.; Feng, K.; et al. Prostate Cancer-associated SPOP mutations enhance cancer cell survival and docetaxel resistance by upregulating Caprin1-dependent stress granule assembly. Mol. Cancer 2019, 18, 170. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Urao, N.; Ashino, T.; Sudhahar, V.; Inomata, H.; Yamaoka-Tojo, M.; McKinney, R.D.; Fukai, T.; Ushio-Fukai, M. IQGAP1 links PDGF receptor-β signal to focal adhesions involved in vascular smooth muscle cell migration: Role in neointimal formation after vascular injury. Am. J. Physiol. Physiol. 2013, 305, C591–C600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Z.L.; Mo, Y.; Yu, Y.J.; Chong, B.H. PDGFR Inhibitor Imatinib May be a Potent Drug in Treating Essential Thrombocythemia. Blood 2016, 128, 5470. [Google Scholar] [CrossRef]
- Rosenberg, A.; Mathew, P. Imatinib and prostate cancer: Lessons learned from targeting the platelet-derived growth factor receptor. Expert Opin. Investig. Drugs 2013, 22, 787–794. [Google Scholar] [CrossRef]
- Kadivar, A.; Kamalidehghan, B.; Javar, H.A.; Karimi, B.; Sedghi, R.; Bin Noordin, M.I. Antiproliferation effect of imatinib mesylate on MCF7, T-47D tumorigenic and MCF 10A nontumorigenic breast cell lines via PDGFR-β, PDGF-BB, c-Kit and SCF genes. Drug Des. Dev. Ther. 2017, 11, 469–481. [Google Scholar] [CrossRef] [Green Version]
- Chott, A.; Sun, Z.; Morganstern, D.; Pan, J.; Li, T.; Susani, M.; Mosberger, I.; Upton, M.P.; Bubley, G.J.; Balk, S.P. Tyrosine Kinases Expressed in Vivo by Human Prostate Cancer Bone Marrow Metastases and Loss of the Type 1 Insulin-Like Growth Factor Receptor. Am. J. Pathol. 1999, 155, 1271–1279. [Google Scholar] [CrossRef]
- Frolov, A.; Chahwan, S.; Ochs, M.; Arnoletti, J.P.; Pan, Z.-Z.; Favorova, O.; Fletcher, J.; Von Mehren, M.; Eisenberg, B.; Godwin, A.K. Response markers and the molecular mechanisms of action of Gleevec in gastrointestinal stromal tumors. Mol. Cancer Ther. 2003, 2, 699–709. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| SLC45A3—ERG-Negative | SLC45A3—ERG-Positive | p-Value | |
|---|---|---|---|
| (N = 50) | (N = 10) | ||
| Age | 61.6 ± 7.4 | 58.7 ± 7.7 | 0.269 |
| M stage | 1 | ||
| - M0 | 45 (97.8%) | 8 (100.0%) | |
| - M1b | 1 (2.2%) | 0 (0.0%) | |
| T stage | 0.884 | ||
| - T1c | 19 (46.3%) | 4 (66.7%) | |
| - T2a | 5 (12.2%) | 1 (16.7%) | |
| - T2b | 4 (9.8%) | 0 (0.0%) | |
| - T2c | 4 (9.8%) | 0 (0.0%) | |
| - T3a | 5 (12.2%) | 1 (16.7%) | |
| - T3b | 3 (7.3%) | 0 (0.0%) | |
| - T4 | 1 (2.4%) | 0 (0.0%) | |
| Gleason Score | 0.474 | ||
| - 6 | 6 (12.0%) | 2 (20.0%) | |
| - 7 | 25 (50.0%) | 7 (70.0%) | |
| - 8 | 7 (14.0%) | 1 (10.0%) | |
| - 9 | 11 (22.0%) | 0 (0.0%) | |
| - 10 | 1 (2.0%) | 0 (0.0%) | |
| Laterality | 0.324 | ||
| - Bilateral | 43 (86.0%) | 7 (70.0%) | |
| - Left | 1 (2.0%) | 0 (0.0%) | |
| - Right | 6 (12.0%) | 3 (30.0%) | |
| Vital Status | 1 | ||
| - LIVING | 48 (96.0%) | 10 (100.0%) | |
| - DECEASED | 2 (4.0%) | 0 (0.0%) | |
| PSA level | 9.7 ± 52.3 | 0.1 ± 0.0 | 0.262 |
| Race | |||
| - WHITE | 9 (100.0%) | 4 (100.0%) |
| Target Gene List | Actionable Drugs | Related Pathway |
|---|---|---|
| CSF1R | GW-2580, Imatinib | Focal adhesion kinase (FAK)/Pl3k-Akt signaling pathway, |
| Receptor tyrosine kinase/PDGF signaling pathway | ||
| ESR1 | Fulvestrant, Tamoxifen, Letrozole, Palbociclib | Receptor tyrosine kinase/PDGF signaling pathway |
| FOS | Irbesartan | TGF-beta signaling pathway |
| HGF | MET tyrosine kinase inhibitor SGX523 | Focal adhesion kinase (FAK)/Pl3k-Akt signaling pathway, |
| Receptor tyrosine kinase/PDGF signaling pathway | ||
| JUN | Irbesartan | Focal adhesion kinase (FAK)/Pl3k-Akt signaling pathway, |
| VEGFA signaling pathway, Wnt signaling pathway, TGF-beta signaling pathway | ||
| PDGFRA | Crenolanib, Imatinib, Sunitinib, Sorafenib, Ponatinib | Focal adhesion kinase (FAK)/Pl3k-Akt signaling pathway, |
| Receptor tyrosine kinase/PDGF signaling pathway, JAK STAT signaling pathway | ||
| PDGFRB | Sunitinib, Dasatinib, Imatinib, Chemotherapy | Focal adhesion kinase (FAK)/Pl3k-Akt signaling pathway, |
| JAK STAT signaling pathway, Receptor tyrosine kinase/PDGF signaling pathway | ||
| PTGS2 | Aspirin | VEGFA signaling pathway |
| ZEB1 | Doxorubicin, Salinomycin | TGF-beta signaling pathway |
| ABCB1 | Paclitaxel, Cisplatin, Carboplatin | NA |
| DDR2 | Dasatinib, Erlotinib | NA |
| DPYD | Leucovorin, Fluorouracil | NA |
| NT5E | Cetuximab | NA |
| PTPRB | Sunitinib | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Hwang, K.W.; Lee, H.J.; Kim, H.S. Systematic Analysis of Cellular Signaling Pathways and Therapeutic Targets for SLC45A3:ERG Fusion-Positive Prostate Cancer. J. Pers. Med. 2022, 12, 1818. https://doi.org/10.3390/jpm12111818
Kim J, Hwang KW, Lee HJ, Kim HS. Systematic Analysis of Cellular Signaling Pathways and Therapeutic Targets for SLC45A3:ERG Fusion-Positive Prostate Cancer. Journal of Personalized Medicine. 2022; 12(11):1818. https://doi.org/10.3390/jpm12111818
Chicago/Turabian StyleKim, Jongsu, Kyung Won Hwang, Hye Jung Lee, and Hong Sook Kim. 2022. "Systematic Analysis of Cellular Signaling Pathways and Therapeutic Targets for SLC45A3:ERG Fusion-Positive Prostate Cancer" Journal of Personalized Medicine 12, no. 11: 1818. https://doi.org/10.3390/jpm12111818