
Cytokine Pathways in Cardiac Dysfunction following Burn Injury and Changes in Genome Expression


Abstract
:1. Introduction
2. The Role of Cytokines in Burn-Induced Cardiac Dysfunction
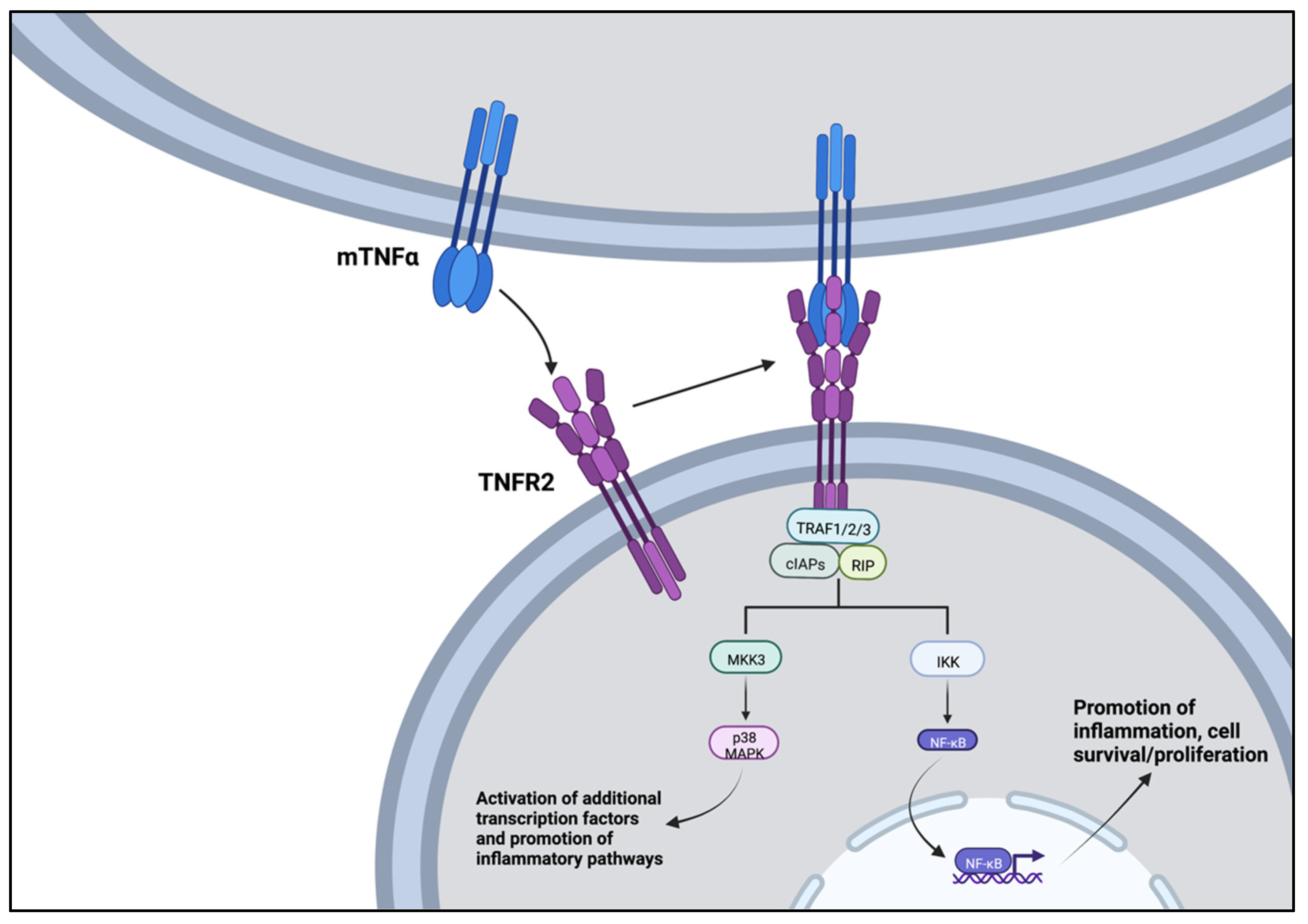
2.1. Tumor Necrosis Factor α (TNFα)
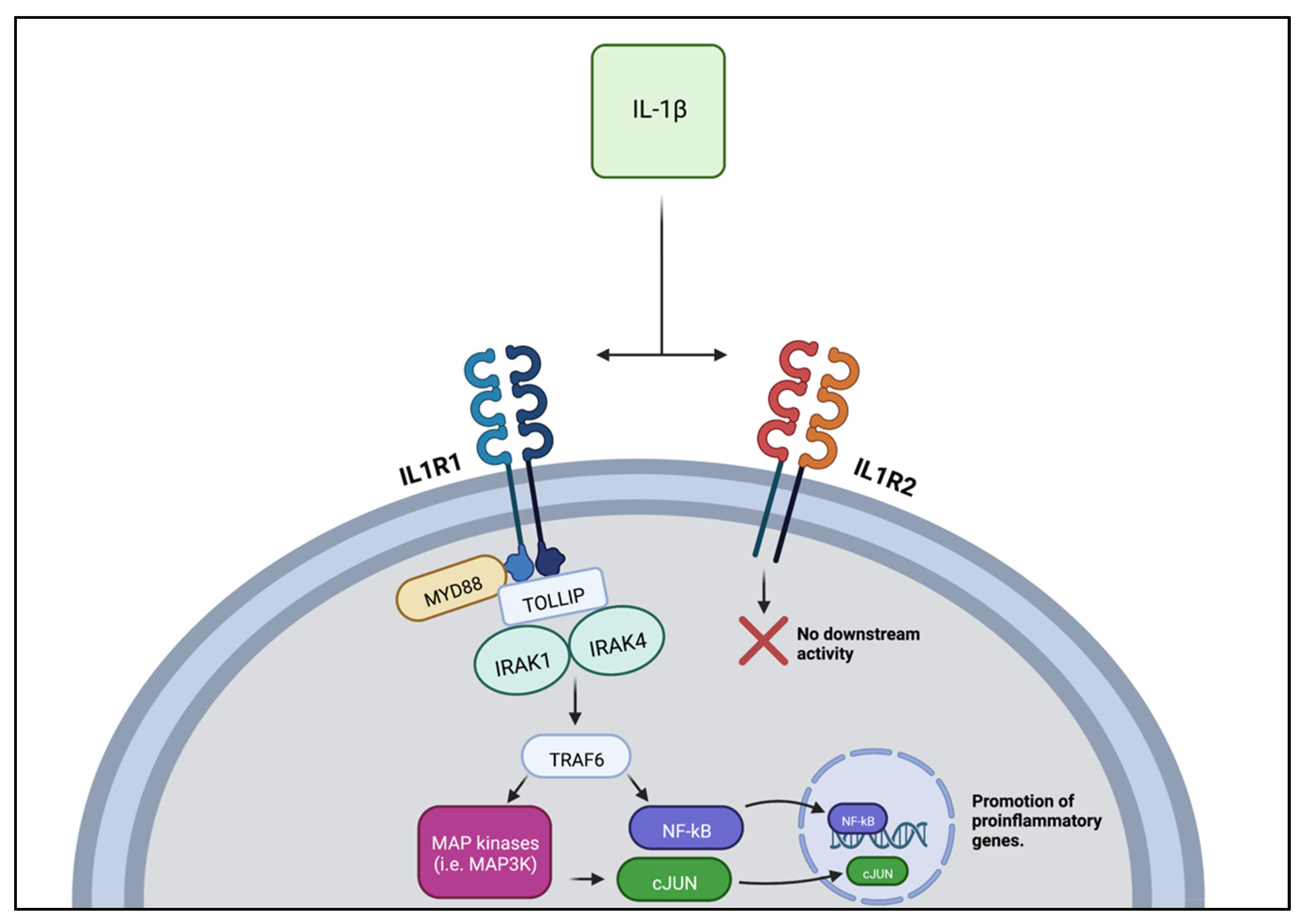
2.2. Interleukin 1 β (IL-1β)
2.3. Interleukin-6 (IL-6)
2.4. Interleukin-10 (IL-10)
3. Changes in the Cytokine Transcriptome in Inflammation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Burn Incidence Fact Sheet—American Burn Association. Available online: https://ameriburn.org/who-we-are/media/burn-incidence-fact-sheet/ (accessed on 8 September 2022).
- Adams, H.R.; Baxter, C.R.; Izenberg, S.D. Decreased Contractility and Compliance of the Left Ventricle as Complications of Thermal Trauma. Am. Heart J. 1984, 108, 1477–1487. [Google Scholar] [CrossRef]
- Ballard-Croft, C.; White, D.J.; Maass, D.L.; Hybki, D.P.; Horton, J.W. Role of P38 Mitogen-Activated Protein Kinase in Cardiac Myocyte Secretion of the Inflammatory Cytokine TNF-α. Am. J. Physiol.-Heart Circ. Physiol. 2001, 280, H1970–H1981. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.S.; Hermann, D.G.; McQuitty, A.L.; Woodson, L.C.; Kramer, G.C.; Herndon, D.N.; Ford, P.M.; Kinsky, M.P. Burn-Induced Cardiac Dysfunction Increases Length of Stay in Pediatric Burn Patients. J. Burn Care Res. 2013, 34, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.T.; Barrow, R.E.; Sterns, A.M.; Hawkins, H.K.; Kimbrough, C.W.; Jeschke, M.G.; Lee, J.O.; Sanford, A.P.; Herndon, D.N. Age-Dependent Differences in Survival after Severe Burns: A Unicentric Review of 1674 Patients and 179 Autopsies over 15 Years. J. Am. Coll. Surg. 2006, 202, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Mak, G.Z.; Hardy, A.R.; Meyer, R.A.; Kagan, R.J. Reversible Cardiomyopathy After Severe Burn Injury. J. Burn Care Res. 2006, 27, 482–486. [Google Scholar] [CrossRef]
- Williams, F.N.; Herndon, D.N.; Suman, O.E.; Lee, J.O.; Norbury, W.B.; Branski, L.K.; Mlcak, R.P.; Jeschke, M.G. Changes in cardiac physiology after severe burn injury. J. Burn. Care Res. 2011, 32, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Calvano, S.E.; Xiao, W.; Richards, D.R.; Felciano, R.M.; Baker, H.V.; Cho, R.J.; Chen, R.O.; Brownstein, B.H.; Cobb, J.P.; Tschoeke, S.K.; et al. A Network-Based Analysis of Systemic Inflammation in Humans. Nature 2005, 437, 1032–1037. [Google Scholar] [CrossRef]
- Desai, K.H.; Tan, C.S.; Leek, J.T.; Maier, R.V.; Tompkins, R.G.; Storey, J.D.; the Inflammation and the Host Response to Injury Large-Scale Collaborative Research Program. Dissecting Inflammatory Complications in Critically Injured Patients by Within-Patient Gene Expression Changes: A Longitudinal Clinical Genomics Study. PLoS Med. 2011, 8, e1001093. [Google Scholar] [CrossRef] [Green Version]
- Keyloun, J.W.; Campbell, R.; Carney, B.C.; Yang, R.; Miller, S.-A.; Detwiler, L.; Gautam, A.; Moffatt, L.T.; Hammamieh, R.; Jett, M.; et al. Early Transcriptomic Response to Burn Injury: Severe Burns Are Associated With Immune Pathway Shutdown. J. Burn Care Res. 2022, 43, 306–314. [Google Scholar] [CrossRef]
- Xiao, W.; Mindrinos, M.N.; Seok, J.; Cuschieri, J.; Cuenca, A.G.; Gao, H.; Hayden, D.L.; Hennessy, L.; Moore, E.E.; Minei, J.P.; et al. A Genomic Storm in Critically Injured Humans. J. Exp. Med. 2011, 208, 2581–2590. [Google Scholar] [CrossRef]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front. Immunol. 2021, 11, 594150. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D. Cytokine-Induced Modulation of Cardiac Function. Circ. Res. 2004, 95, 1140–1153. [Google Scholar] [CrossRef]
- Weber, B.; Lackner, I.; Gebhard, F.; Miclau, T.; Kalbitz, M. Trauma, a Matter of the Heart—Molecular Mechanism of Post-Traumatic Cardiac Dysfunction. Int. J. Mol. Sci. 2021, 22, 737. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, C.; Wang, D.W. Inflammatory Cytokines, Immune Cells, and Organ Interactions in Heart Failure. Front. Physiol. 2021, 12, 903. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Frangogiannis, N.G. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863. [Google Scholar] [CrossRef]
- Drexler, H. Nitric Oxide Synthases in the Failing Human Heart. Circulation 1999, 99, 2972–2975. [Google Scholar] [CrossRef] [Green Version]
- Duncan, D.J.; Hopkins, P.M.; Harrison, S.M. Negative Inotropic Effects of Tumour Necrosis Factor-Alpha and Interleukin-1beta Are Ameliorated by Alfentanil in Rat Ventricular Myocytes. Br. J. Pharmacol. 2007, 150, 720–726. [Google Scholar] [CrossRef] [Green Version]
- Duncan, D.J.; Yang, Z.; Hopkins, P.M.; Steele, D.S.; Harrison, S.M. TNF-α and IL-1β Increase Ca2+ Leak from the Sarcoplasmic Reticulum and Susceptibility to Arrhythmia in Rat Ventricular Myocytes. Cell Calcium 2010, 47, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, T.; Vaca, L.; Rossen, R.D.; Durante, W.; Hazarika, P.; Mann, D.L. Cellular Basis for the Negative Inotropic Effects of Tumor Necrosis Factor-a in the Adult Mammalian Heart. J. Clin. Investig. 1993, 92, 2303–2312. [Google Scholar]
- Braz, J.C.; Gregory, K.; Pathak, A.; Zhao, W.; Sahin, B.; Klevitsky, R.; Kimball, T.F.; Lorenz, J.N.; Nairn, A.C.; Liggett, S.B.; et al. PKC-α Regulates Cardiac Contractility and Propensity toward Heart Failure. Nat. Med. 2004, 10, 248–254. [Google Scholar] [CrossRef]
- Jude, B.; Vetel, S.; Giroux-Metges, M.A.; Pennec, J.P. Rapid Negative Inotropic Effect Induced by TNF-α in Rat Heart Perfused Related to PKC Activation. Cytokine 2018, 107, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Maass, D.L.; White, D.J.; Horton, J.W. Effects of Burn Injury on Myocardial Signaling and Cytokine Secretion: Possible Role of PKC. Am. J. Physiol-Regul. Integr. Comp. Physiol. 2007, 292, R887–R896. [Google Scholar] [CrossRef] [PubMed]
- Giroir, B.P.; Horton, J.W.; White, D.J.; McIntyre, K.L.; Lin, C.Q. Inhibition of Tumor Necrosis Factor Prevents Myocardial Dysfunction during Burn Shock. Am. J. Physiol. 1994, 267, H118–H124. [Google Scholar] [CrossRef]
- Liu, S.J.; Zhou, W.; Kennedy, R.H. Suppression of β-Adrenergic Responsiveness of L-Type Ca2+ Current by IL-1β in Rat Ventricular Myocytes. Am. J. Physiol.-Heart Circ. Physiol. 1999, 276, H141–H148. [Google Scholar] [CrossRef] [PubMed]
- Grothusen, C.; Hagemann, A.; Attmann, T.; Braesen, J.; Broch, O.; Cremer, J.; Schoettler, J. Impact of an Interleukin-1 Receptor Antagonist and Erythropoietin on Experimental Myocardial Ischemia/Reperfusion Injury. Sci. World J. 2012, 2012, e737585. [Google Scholar] [CrossRef] [Green Version]
- Zuo, S.; Li, L.; Ruan, Y.; Jiang, L.; Li, X.; Li, S.; Wen, S.; Bai, R.; Liu, N.; Du, X.; et al. Acute Administration of Tumour Necrosis Factor-α Induces Spontaneous Calcium Release via the Reactive Oxygen Species Pathway in Atrial Myocytes. EP Eur. 2018, 20, 1367–1374. [Google Scholar] [CrossRef]
- Mann, D.L.; McMurray, J.J.V.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted Anticytokine Therapy in Patients With Chronic Heart Failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a Single Dose of the Interleukin-6 Receptor Antagonist Tocilizumab on Inflammation and Troponin T Release in Patients with Non-ST-Elevation Myocardial Infarction: A Double-Blind, Randomized, Placebo-Controlled Phase 2 Trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef] [Green Version]
- Sanin, D.E.; Prendergast, C.T.; Mountford, A.P. IL-10 Production in Macrophages Is Regulated by a TLR-Driven CREB-Mediated Mechanism That Is Linked to Genes Involved in Cell Metabolism. J. Immunol. 2015, 195, 1218–1232. [Google Scholar] [CrossRef] [Green Version]
- Casale, G.P.; Thompson, J.R.; Carpenter, L.C.; Kim, J.; Lackner, T.J.; Mietus, C.J.; Ha, D.M.; Myers, S.A.; Brunette, K.E.; Li, S.; et al. Cytokine Signature of Inflammation Mediated by Autoreactive Th-Cells, in Calf Muscle of Claudicating Patients with Fontaine Stage II Peripheral Artery Disease. Transl. Res. 2021, 228, 94–108. [Google Scholar] [CrossRef]
- Cornick, S.M.; de Noronha, S.A.A.C.; de Noronha, S.M.R.; Cezillo, M.V.B.; Ferreira, L.M.; Gragnani, A. Toll like Receptors Gene Expression of Human Keratinocytes Cultured of Severe Burn Injury. Acta Cir. Bras. 2014, 29, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.-S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The Role of Reactive Oxygen Species in the Pathophysiology of Cardiovascular Diseases and the Clinical Significance of Myocardial Redox. Ann. Transl. Med. 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, L.H.; Bhavsar, D.; Mailänder, P. The Biology of Burn Injury. Exp. Dermatol. 2010, 19, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, S.M.; Naga Prasad, S.V. Tumor Necrosis Factor-α in Heart Failure: An Updated Review. Curr. Cardiol. Rep. 2018, 20, 117. [Google Scholar] [CrossRef]
- Jang, D.; Lee, A.-H.; Shin, H.-Y.; Song, H.-R.; Park, J.-H.; Kang, T.-B.; Lee, S.-R.; Yang, S.-H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Mann, D.L.; Young, J.B. Basic Mechanisms in Congestive Heart Failure: Recognizing the Role of Proinflammatory Cytokines. Chest 1994, 105, 897–904. [Google Scholar] [CrossRef]
- Pagani, F.D.; Baker, L.S.; Hsi, C.; Knox, M.; Fink, M.P.; Visner, M.S. Left Ventricular Systolic and Diastolic Dysfunction after Infusion of Tumor Necrosis Factor-Alpha in Conscious Dogs. J. Clin. Investig. 1992, 90, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Finkel, M.S.; Oddis, C.V.; Jacob, T.D.; Watkins, S.C.; Hattler, B.G.; Simmons, R.L. Negative Inotropic Effects of Cytokines on the Heart Mediated by Nitric Oxide. Science 1992, 257, 387–389. [Google Scholar] [CrossRef]
- Eichenholz, P.W.; Eichacker, P.Q.; Hoffman, W.D.; Banks, S.M.; Parrillo, J.E.; Danner, R.L.; Natanson, C. Tumor Necrosis Factor Challenges in Canines: Patterns of Cardiovascular Dysfunction. Am. J. Physiol. 1992, 263, H668–H675. [Google Scholar] [CrossRef]
- Tracey, K.J.; Beutler, B.; Lowry, S.F.; Merryweather, J.; Wolpe, S.; Milsark, I.W.; Hariri, R.J.; Fahey, T.J.; Zentella, A.; Albert, J.D. Shock and Tissue Injury Induced by Recombinant Human Cachectin. Science 1986, 234, 470–474. [Google Scholar] [CrossRef]
- Wen, J.J.; Williams, T.P.; Cummins, C.B.; Colvill, K.M.; Radhakrishnan, G.L.; Radhakrishnan, R.S. Effect of Mitochondrial Antioxidant (Mito-TEMPO) on Burn-Induced Cardiac Dysfunction. J. Am. Coll. Surg. 2021, 232, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Maass, D.L.; Hybki, D.P.; White, J.; Horton, J.W. The Time Course of Cardiac NF-ΚB Activation and TNF-α Secretion by Cardiac Myocytes After Burn Injury: Contribution to Burn-Related Cardiac Contractile Dysfunction. Shock 2002, 17, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.J.; Mobli, K.; Radhakrishnan, G.L.; Radhakrishnan, R.S. Regulation of Key Immune-Related Genes in the Heart Following Burn Injury. J. Pers. Med. 2022, 12, 1007. [Google Scholar] [CrossRef] [PubMed]
- Giroir, B.P.; Johnson, J.H.; Brown, T.; Allen, G.L.; Beutler, B. The Tissue Distribution of Tumor Necrosis Factor Biosynthesis during Endotoxemia. J. Clin. Investig. 1992, 90, 693–698. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Van Remmen, H. The SarcoEndoplasmic Reticulum Calcium ATPase (SERCA) Pump: A Potential Target for Intervention in Aging and Skeletal Muscle Pathologies. Skelet. Muscle 2021, 11, 25. [Google Scholar] [CrossRef]
- Umar, S.; van der Laarse, A. Nitric Oxide and Nitric Oxide Synthase Isoforms in the Normal, Hypertrophic, and Failing Heart. Mol. Cell Biochem. 2009, 333, 191. [Google Scholar] [CrossRef]
- Deswal, A.; Bozkurt, B.; Seta, Y.; Parilti-Eiswirth, S.; Hayes, F.A.; Blosch, C.; Mann, D.L. Safety and Efficacy of a Soluble P75 Tumor Necrosis Factor Receptor (Enbrel, Etanercept) in Patients With Advanced Heart Failure. Circulation 1999, 99, 3224–3226. [Google Scholar] [CrossRef]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Randomized, Double-Blind, Placebo-Controlled, Pilot Trial of Infliximab, a Chimeric Monoclonal Antibody to Tumor Necrosis Factor-α, in Patients With Moderate-to-Severe Heart Failure. Circulation 2003, 107, 3133–3140. [Google Scholar] [CrossRef] [Green Version]
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural Basis of IL-1 Family Cytokine Signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef] [Green Version]
- Gulick, T.; Chung, M.K.; Pieper, S.J.; Lange, L.G.; Schreiner, G.F. Interleukin 1 and Tumor Necrosis Factor Inhibit Cardiac Myocyte Beta-Adrenergic Responsiveness. Proc. Natl. Acad. Sci. USA 1989, 86, 6753–6757. [Google Scholar] [CrossRef] [Green Version]
- Van Tassell, B.W.; Seropian, I.M.; Toldo, S.; Mezzaroma, E.; Abbate, A. Interleukin-1β Induces a Reversible Cardiomyopathy in the Mouse. Inflamm. Res. 2013, 62, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Matarrese, P.; Maccari, S.; Vona, R.; Gambardella, L.; Stati, T.; Marano, G. Role of β-Adrenergic Receptors and Estrogen in Cardiac Repair after Myocardial Infarction: An Overview. Int. J. Mol. Sci. 2021, 22, 8957. [Google Scholar] [CrossRef] [PubMed]
- Combes, A.; Frye, C.; Lemster, B.; Brooks, S.; Watkins, S.; Feldman, A.; McTiernan, C. Chronic Exposure to Interleukin 1β Induces a Delayed and Reversible Alteration in Excitation-Contraction Coupling of Cultured Cardiomyocytes. Pflügers Arch. 2002, 445, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.J.; Cummins, C.B.; Radhakrishnan, R.S. Burn-Induced Cardiac Mitochondrial Dysfunction via Interruption of the PDE5A-CGMP-PKG Pathway. Int. J. Mol. Sci. 2020, 21, 2350. [Google Scholar] [CrossRef] [Green Version]
- Sierawska, O.; Małkowska, P.; Taskin, C.; Hrynkiewicz, R.; Mertowska, P.; Grywalska, E.; Korzeniowski, T.; Torres, K.; Surowiecka, A.; Niedźwiedzka-Rystwej, P.; et al. Innate Immune System Response to Burn Damage-Focus on Cytokine Alteration. Int. J. Mol. Sci. 2022, 23, 716. [Google Scholar] [CrossRef]
- Toldo, S.; Schatz, A.M.; Mezzaroma, E.; Chawla, R.; Stallard, T.W.; Stallard, W.C.; Jahangiri, A.; Van Tassell, B.W.; Abbate, A. Recombinant Human Interleukin-1 Receptor Antagonist Provides Cardioprotection During Myocardial Ischemia Reperfusion in the Mouse. Cardiovasc. Drugs Ther. 2012, 26, 273–276. [Google Scholar] [CrossRef]
- Wen, J.J.; Mobli, K.; Rontoyanni, V.G.; Cummins, C.B.; Radhakrishnan, G.L.; Murton, A.; Radhakrishnan, R.S. Nuclear Factor Erythroid 2-Related Factor 2 Activation and Burn-Induced Cardiac Dysfunction. J. Am. Coll. Surg. 2022, 234, 660–671. [Google Scholar] [CrossRef]
- Song, M.; Kellum, J.A. Interleukin-6. Crit. Care Med. 2005, 33, S463. [Google Scholar] [CrossRef]
- Van Snick, J. Interleukin-6: An Overview. Annu. Rev. Immunol. 1990, 8, 253–278. [Google Scholar] [CrossRef]
- Kraft, R.; Herndon, D.N.; Finnerty, C.C.; Cox, R.A.; Song, J.; Jeschke, M.G. Predictive Value of IL-8 for Sepsis and Severe Infections after Burn Injury-A Clinical Study. Shock 2015, 43, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, H.; Matsumoto, H.; Osuka, A.; Ogura, H.; Shimizu, K.; Kang, S.; Tanaka, T.; Ueyama, M.; Shimazu, T. Clinical Importance of a Cytokine Network in Major Burns. Shock 2019, 51, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, B.; Mitchell, D.H.; Colston, J.T.; Freeman, G.L. Regulation of CCAAT/Enhancer Binding Protein, Interleukin-6, Interleukin-6 Receptor, and Gp130 Expression during Myocardial Ischemia/Reperfusion. Circulation 1999, 99, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Högye, M.; Mándi, Y.; Csanády, M.; Sepp, R.; Buzás, K. Comparison of Circulating Levels of Interleukin-6 and Tumor Necrosis Factor-Alpha in Hypertrophic Cardiomyopathy and in Idiopathic Dilated Cardiomyopathy. Am. J. Cardiol. 2004, 94, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Parissis, J.T.; Adamopoulos, S.N.; Venetsanou, K.F.; Karas, S.M.; Kremastinos, D.T. Elevated Plasma Amylase Levels in Advanced Chronic Heart Failure Secondary to Ischemic or Idiopathic Dilated Cardiomyopathy: Correlation with Circulating Interleukin-6 Activity. J. Interferon Cytokine Res. 2003, 23, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.J.; Liu, X.L.; He, G.X.; Xu, H.P. Effects of Single-Dose Atorvastatin on Interleukin-6, Interferon Gamma, and Myocardial No-Reflow in a Rabbit Model of Acute Myocardial Infarction and Reperfusion. Braz. J. Med. Biol. Res. 2014, 47, 245–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, H.-Y.; Bassel-Duby, R.; Maass, D.L.; Johnston, W.E.; Horton, J.W.; Tao, W. Role of Interleukin-6 in Cardiac Inflammation and Dysfunction after Burn Complicated by Sepsis. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H2408–H2416. [Google Scholar] [CrossRef] [Green Version]
- Ballard-Croft, C.; Carlson, D.; Maass, D.L.; Horton, J.W. Burn Trauma Alters Calcium Transporter Protein Expression in the Heart. J. Appl. Physiol. 2004, 97, 1470–1476. [Google Scholar] [CrossRef] [Green Version]
- Villegas, S.; Villarreal, F.J.; Dillmann, W.H. Leukemia Inhibitory Factor and Interleukin-6 Downregulate Sarcoplasmic Reticulum Ca2+ ATPase (SERCA2) in Cardiac Myocytes. Basic Res. Cardiol. 2000, 95, 47–54. [Google Scholar] [CrossRef]
- Yu, X.; Kennedy, R.H.; Liu, S.J. JAK2/STAT3, Not ERK1/2, Mediates Interleukin-6-Induced Activation of Inducible Nitric-Oxide Synthase and Decrease in Contractility of Adult Ventricular Myocytes. J. Biol. Chem. 2003, 278, 16304–16309. [Google Scholar] [CrossRef] [Green Version]
- Broch, K.; Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Tøllefsen, I.M.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damås, J.K.; et al. Randomized Trial of Interleukin-6 Receptor Inhibition in Patients With Acute ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2021, 77, 1845–1855. [Google Scholar] [CrossRef]
- Llopiz, D.; Ruiz, M.; Infante, S.; Villanueva, L.; Silva, L.; Hervas-Stubbs, S.; Alignani, D.; Guruceaga, E.; Lasarte, J.J.; Sarobe, P. IL-10 Expression Defines an Immunosuppressive Dendritic Cell Population Induced by Antitumor Therapeutic Vaccination. Oncotarget 2016, 8, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Marra, L.E.; Zhang, Z.X.; Joe, B.; Campbell, J.; Levy, G.A.; Penninger, J.; Zhang, L. IL-10 Induces Regulatory T Cell Apoptosis by Up-Regulation of the Membrane Form of TNF-α. J. Immunol. 2004, 172, 1028–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsurumi, A.; Que, Y.-A.; Ryan, C.M.; Tompkins, R.G.; Rahme, L.G. TNF-α/IL-10 Ratio Correlates with Burn Severity and May Serve as a Risk Predictor of Increased Susceptibility to Infections. Front. Public Health 2016, 4, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Liu, L.; Zhang, X.; Fan, X.; Krishnamurthy, P.; Verma, S.; Tongers, J.; Misener, S.; Ashcherkin, N.; Sun, H.; et al. IL-10 Provides Cardioprotection in Diabetic Myocardial Infarction via Upregulation of Heme Clearance Pathways. JCI Insight 2020, 5, e133050. [Google Scholar] [CrossRef]
- Verma, S.K.; Krishnamurthy, P.; Barefield, D.; Singh, N.; Gupta, R.; Lambers, E.; Thal, M.; Mackie, A.; Hoxha, E.; Ramirez, V.; et al. Interleukin-10 Treatment Attenuates Pressure Overload–Induced Hypertrophic Remodeling and Improves Heart Function via Signal Transducers and Activators of Transcription 3–Dependent Inhibition of Nuclear Factor-ΚB. Circulation 2012, 126, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 Inhibits Inflammation and Attenuates Left Ventricular Remodeling After Myocardial Infarction via Activation of STAT3 and Suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef] [Green Version]
- Lyons, A.; Goebel, A.; Mannick, J.A.; Lederer, J.A. Protective Effects of Early Interleukin 10 Antagonism on Injury-Induced Immune Dysfunction. Arch. Surg. 1999, 134, 1317–1324. [Google Scholar] [CrossRef] [Green Version]
- Pileri, D.; Accardo Palombo, A.; D’Amelio, L.; D’Arpa, N.; Amato, G.; Masellis, A.; Cataldo, V.; Mogavero, R.; Napoli, B.; Lombardo, C.; et al. Concentrations of Cytokines Il-6 and Il-10 in Plasma of Burn Patients: Their Relationship to Sepsis and Outcome. Ann Burn. Fire Disasters 2008, 21, 182–185. [Google Scholar]
- Finnerty, C.C.; Herndon, D.N.; Przkora, R.; Pereira, C.T.; Oliveira, H.M.; Queiroz, D.M.M.; Rocha, A.M.C.; Jeschke, M.G. Cytokine expression profile over time in severely burned pediatric patients. Shock 2006, 26, 13–19. [Google Scholar] [CrossRef]
- Gragnani, A.; Cezillo, M.V.; da Silva, I.D.; de Noronha, S.M.R.; Correa-Noronha, S.A.; Ferreira, L.M. Gene expression profile of cytokines and receptors of inflammation from cultured keratinocytes of burned patients. Burns 2014, 40, 947–956. [Google Scholar] [CrossRef]
- Gragnani, A.; Müller, B.R.; da Silva, I.D.C.G.; de Noronha, S.M.R.; Ferreira, L.M. Keratinocyte Growth Factor, Tumor Necrosis Factor-Alpha and Interleukin-1 Beta Gene Expression in Cultured Fibroblasts and Keratinocytes from Burned Patients. Acta Cir. Bras. 2013, 28, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seys, S.F.; Grabowski, M.; Adriaensen, W.; Decraene, A.; Dilissen, E.; Vanoirbeek, J.A.; Dupont, L.J.; Ceuppens, J.L.; Bullens, D.M.A. Sputum Cytokine Mapping Reveals an “IL-5, IL-17A, IL-25-High” Pattern Associated with Poorly Controlled Asthma. Clin. Exp. Allergy 2013, 43, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Baines, K.J.; Wood, L.G.; Gibson, P.G. Systemic Inflammation Is Associated with Differential Gene Expression and Airway Neutrophilia in Asthma. OMICS A J. Integr. Biol. 2013, 17, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Sánchez, L.; Madrid-Miller, A.; Chávez-Rueda, K.; Legorreta-Haquet, M.V.; Tesoro-Cruz, E.; Blanco-Favela, F. Activation of TLR2 and TLR4 by Minimally Modified Low-Density Lipoprotein in Human Macrophages and Monocytes Triggers the Inflammatory Response. Hum. Immunol. 2010, 71, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; Schiraldi, M.; Uguccioni, M.; Bianchi, M.E. HMGB1 and Leukocyte Migration during Trauma and Sterile Inflammation. Mol. Immunol. 2013, 55, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Angele, M.K.; Schwacha, M.G.; Ayala, A.; Chaudry, I.H. Effect of gender and sex hormones on immune responses following shock. Shock 2000, 14, 81–90. [Google Scholar] [CrossRef]
- McClelland, E.E.; Smith, J.M. Gender Specific Differences in the Immune Response to Infection. Arch. Immunol. Ther. Exp. 2011, 59, 203–213. [Google Scholar] [CrossRef]
- Everhardt Queen, A.; Moerdyk-Schauwecker, M.; McKee, L.M.; Leamy, L.J.; Huet, Y.M. Differential Expression of Inflammatory Cytokines and Stress Genes in Male and Female Mice in Response to a Lipopolysaccharide Challenge. PLoS ONE 2016, 11, e0152289. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Rodríguez, L.; López-Hoyos, M.; Muñoz-Cacho, P.; Martínez-Taboada, V.M. Aging Is Associated with Circulating Cytokine Dysregulation. Cell. Immunol. 2012, 273, 124–132. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Patsouris, D.; Stanojcic, M.; Abdullahi, A.; Rehou, S.; Pinto, R.; Chen, P.; Burnett, M.; Amini-Nik, S. Pathophysiologic Response to Burns in the Elderly. EBioMedicine 2015, 2, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Vanzant, E.L.; Hilton, R.E.; Lopez, C.M.; Zhang, J.; Ungaro, R.F.; Gentile, L.F.; Szpila, B.E.; Maier, R.V.; Cuschieri, J.; Bihorac, A.; et al. Advanced Age Is Associated with Worsened Outcomes and a Unique Genomic Response in Severely Injured Patients with Hemorrhagic Shock. Crit. Care 2015, 19, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreckmann, S.C.; Amini-Nik, S.; Tompkins, R.G.; Vojvodic, M.; Jeschke, M.G. Genome-Wide Comparisons of Gene Expression in Adult versus Elderly Burn Patients. PLoS ONE 2019, 14, e0226425. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Cytokine | Mechanisms in Cardiac Dysfunction | Studied Model | Authors |
|---|---|---|---|
| TNFα |
| Rat, feline cardiomyocytes Rat, mouse cardiomyocytes Rat cardiomyocytes | Duncan et al., 2007 [18] Duncan et al., 2010 [19] Yokoyama et al., 1993 [20] Braz et al., 2004 [21] Jude et al., 2018 [22] Tan et al., 2007 [23] Schulz et al., 1992 [24] |
| IL-1β |
| Rat cardiomyocytes Rat, feline cardiomyocytes Rat, feline cardiomyocytes Rat cardiomyocytes Rat cardiomyocytes | Gulick et al., 1989 [25] Combes et al., 2002 [26] Duncan et al., 2007 [18] Duncan et al., 2010 [19] Yokoyama et al., 1993 [20] Duncan et al., 2007 [18] Duncan et al., 2010 [19] Yokoyama et al., 1993 [20] Zuo et al., 2018 [27] Combes et al., 2002 [26] Wen et al., 2020 [28] |
| IL-6 |
| Mouse cardiomyocytes Hamster papillary muscle | Villegas et al., 2000 [29] Yu et al., 2003 [30] |
| IL-10 |
| Human serum | Lyons et al., 1999 [31] Pileri et al., 2008 [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeJesus, J.E.; Wen, J.J.; Radhakrishnan, R. Cytokine Pathways in Cardiac Dysfunction following Burn Injury and Changes in Genome Expression. J. Pers. Med. 2022, 12, 1876. https://doi.org/10.3390/jpm12111876
DeJesus JE, Wen JJ, Radhakrishnan R. Cytokine Pathways in Cardiac Dysfunction following Burn Injury and Changes in Genome Expression. Journal of Personalized Medicine. 2022; 12(11):1876. https://doi.org/10.3390/jpm12111876
Chicago/Turabian StyleDeJesus, Jana E., Jake J. Wen, and Ravi Radhakrishnan. 2022. "Cytokine Pathways in Cardiac Dysfunction following Burn Injury and Changes in Genome Expression" Journal of Personalized Medicine 12, no. 11: 1876. https://doi.org/10.3390/jpm12111876