
Real-World Results from Combined Screening for Monogenic Genomic Health Risks and Reproductive Risks in 300 Adults


Abstract
:1. Introduction
2. Materials and Methods
2.1. Testing Program and Analysis Tools
2.2. Cohort
2.3. Variant Inclusion
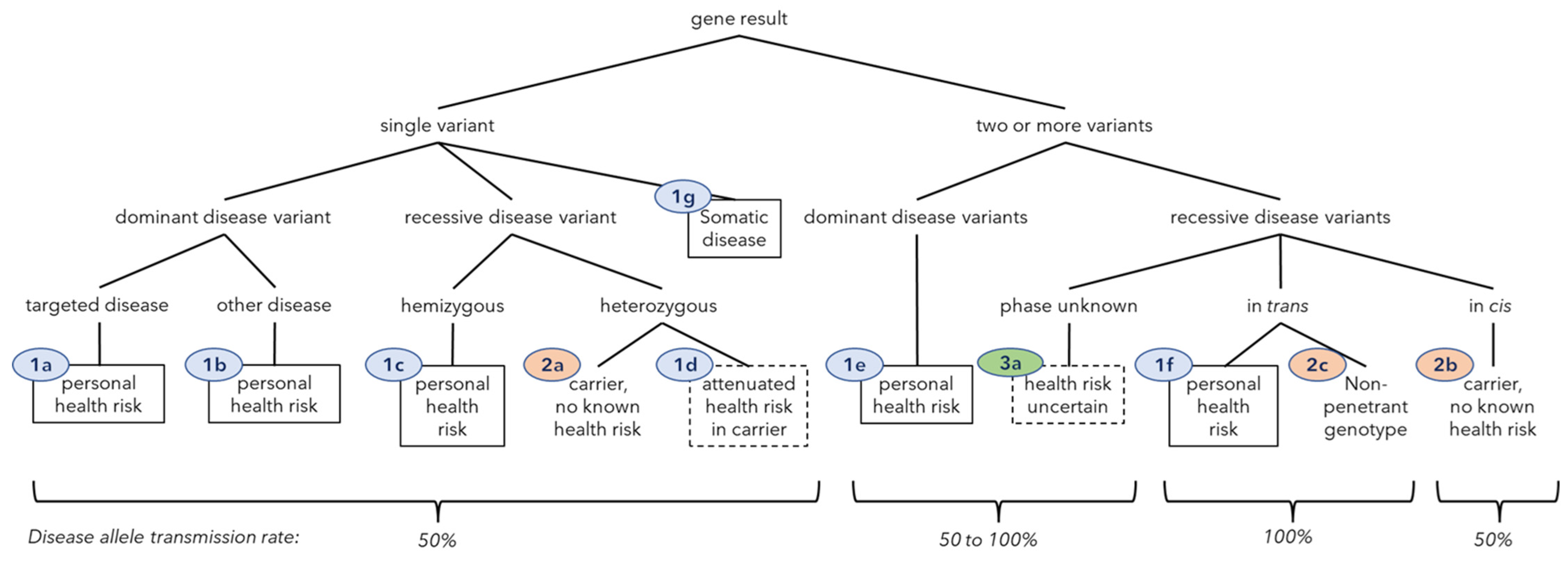
2.4. Gene Result and Clinical Class Definitions
3. Results
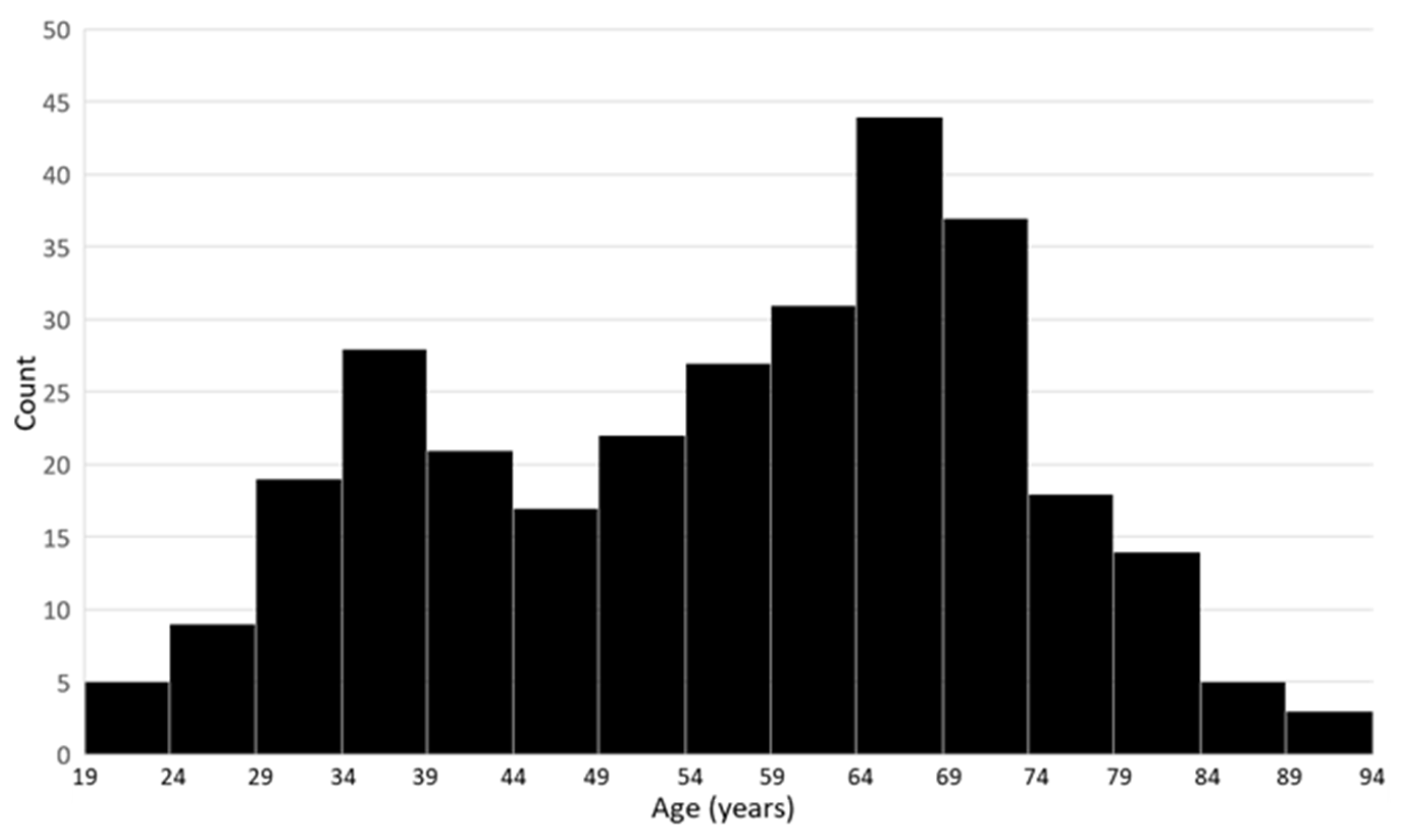
3.1. Cohort Characteristics
3.2. Gene and Variant Prevalence
3.3. Prevalence of Gene Results by Clinical Class
3.3.1. Class 1—Predicted Health Risk
3.3.2. Class 2—Carrier of a Recessive Genetic Disorder, No Known Personal Health Risk
3.3.3. Class 3—Carrier, Health Risk Uncertain
3.4. Overall Frequency of Gene Results with Personal Health Risk and Reproductive Risk Implications
3.5. Comparison to the American College of Medical Genetics and Genomics (ACMG) Secondary Findings Gene Results and Center for Disease Control (CDC) Tier 1 Disorders
4. Discussion
4.1. Cohort Uniqueness
4.2. Cohort Representativeness
4.3. Recessive Carrier Detection
4.4. Personal Health Risk Detection
4.5. Uncertainty and Value
4.6. Gene Result Classification System and Action Plan Messaging
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Cancer and Cardiovascular Risk Variant Tables

{kind=link}
{kind=link}
| Gene | Individuals (n = 300) | Variants |
|---|---|---|
| ATM | 3 | p.Glu522Ilefs*43, p.Arg2034*, p.Leu2463fs (c.7388_7389ins?) |
| BRCA1 | 1 | p.Tyr1853* |
| BRCA2 | 4 | p.Gln1408*, p.Ile2588Phefs*60, p.Glu2846Glyfs*22, p.Gln2859* |
| BRIP1 | 2 | p.Arg798*, Ser895* |
| CHEK2 | 5 | p.Ile157Thr (n = 3), p.Thr367Metfs*15 (n = 2) |
| FH1 | 3 | p.Lys477dup |
| HOXB13 | 1 | p.Gly84Glu |
| MITF | 3 | p.Glu318Lys |
| MSH32 | 1 | deletion (exon 16) |
| MUTYH2 | 8 | p.Tyr179Cys (n = 3), p.Arg274Gln (n = 2), p.Gly396Asp (n = 4), |
| NBN | 1 | p.Lys233Serfs*5 |
| NF1 | 1 | p.Leu1183Arg (possibly mosaic) |
| NTHL12 | 1 | p.Gln90* |
| PMS2 | 1 | p.Lys706Serfs*19 |
| PRKAR1A | 1 | c.709-7_709-2del |
| SDHD | 1 | p.Pro81Leu |
| TP53 | 1 | p.Pro151Arg (possibly mosaic) |
| Gene | Individuals (n = 300) | Variants |
|---|---|---|
| APOB1 | 1 | p.Phe2656Thrfs*10 |
| CAV3 | 1 | p.Ala93Thr |
| DMD2 | 1 | p.Val2159Serfs*4 |
| DSC2 | 1 | c.631-2A>G (splice acceptor) |
| DSG2 | 2 | c.523+1G>A (splice donor), p.Glu1020Alafs*18 |
| F23 | 5 | c.*97G>A (also known as c.20210G>A) |
| F5 | 14 | p.Arg534Gln (also known as “Leiden”) |
| FLNC4 | 1 | deleted gene |
| LDLR | 1 | c.2547+1G>A (splice donor) |
| LMNA | 1 | p.Thr655Asnfs*49 |
| MYBPC3 | 2 | c.3628-41_3628-17del (Intronic), p.Glu542Gln (missense and splice) |
| MYH7 | 1 | p.Lys865Arg |
| PKP2 | 1 | p.Thr50Serfs*61 |
| SERPINC13 | 2 | p.Pro73Leu |
References
- Gregg, A.R.; Aarabi, M.; Klugman, S.; Leach, N.T.; Bashford, M.T.; Goldwaser, T.; Chen, E.; Sparks, T.N.; Reddi, H.V.; Rajkovic, A.; et al. Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: A practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1793–1806. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, S.E. Carrier screening for recessive disorders. Nat. Rev. Genet. 2019, 20, 549–561. [Google Scholar] [CrossRef] [PubMed]
- United_States_Preventative_Services_Task_Force. USPSTF Published Recommendations. Available online: https://uspreventiveservicestaskforce.org/uspstf/topic_search_results?topic_status=P (accessed on 28 September 2022).
- Viera, A.J. Overview of Preventative Care in Adults. UpToDate2022. Available online: https://www.uptodate.com/contents/overview-of-preventive-care-in-adults (accessed on 28 September 2022).
- National_Comprehensive_Cancer_Network. NCCN Guidelines: Detection, Prevention, and Risk Reduction. Available online: https://www.nccn.org/guidelines/category_2 (accessed on 28 September 2022).
- Centers_for_Disease_Control_and_Prevention. Newborn Screening Portal. Available online: https://www.cdc.gov/newbornscreening/index.html (accessed on 28 September 2022).
- Kelly, N.R. Screening Tests in Children and Adolescents. Available online: https://www-uptodate-com/contents/screening-tests-in-children-and-adolescents (accessed on 28 September 2022).
- United_States_Preventative_Services_Task_Force. USPSTF A & B Recommendations. Available online: https://uspreventiveservicestaskforce.org/uspstf/recommendation-topics/uspstf-a-and-b-recommendations (accessed on 28 September 2022).
- Wildin, R.S.; Messersmith, D.J.; Houwink, E.J.F. Modernizing family health history: Achievable strategies to reduce implementation gaps. J. Community Genet. 2021, 12, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Stanich, P.P.; Pelstring, K.R.; Hampel, H.; Pearlman, R. A High Percentage of Early-age Onset Colorectal Cancer Is Potentially Preventable. Gastroenterology 2021, 160, 1850–1852. [Google Scholar] [CrossRef] [PubMed]
- Blout Zawatsky, C.L.; Shah, N.; Machini, K.; Perez, E.; Christensen, K.D.; Zouk, H.; Steeves, M.; Koch, C.; Uveges, M.; Shea, J.; et al. Returning actionable genomic results in a research biobank: Analytic validity, clinical implementation, and resource utilization. Am. J. Hum. Genet. 2021, 108, 2224–2237. [Google Scholar] [CrossRef]
- Gupta, S.; Bharti, B.; Ahnen, D.J.; Buchanan, D.D.; Cheng, I.C.; Cotterchio, M.; Figueiredo, J.C.; Gallinger, S.J.; Haile, R.W.; Jenkins, M.A.; et al. Potential impact of family history-based screening guidelines on the detection of early-onset colorectal cancer. Cancer 2020, 126, 3013–3020. [Google Scholar] [CrossRef]
- Srinivasan, S.; Won, N.Y.; Dotson, W.D.; Wright, S.T.; Roberts, M.C. Barriers and facilitators for cascade testing in genetic conditions: A systematic review. Eur. J. Hum. Genet. 2020, 28, 1631–1644. [Google Scholar] [CrossRef]
- Wildin, R.S.; Giummo, C.A.; Reiter, A.W.; Peterson, T.C.; Leonard, D.G.B. Primary Care Implementation of Genomic Population Health Screening Using a Large Gene Sequencing Panel. Front. Genet. 2022, 13, 867334. [Google Scholar] [CrossRef]
- Haverfield, E.V.; Esplin, E.D.; Aguilar, S.J.; Hatchell, K.E.; Ormond, K.E.; Hanson-Kahn, A.; Atwal, P.S.; Macklin-Mantia, S.; Hines, S.; Sak, C.W.; et al. Physician-directed genetic screening to evaluate personal risk for medically actionable disorders: A large multi-center cohort study. BMC Med. 2021, 19, 199. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef]
- World_Health_Organization. Women of Reproductive Age. Available online: https://www.who.int/data/gho/indicator-metadata-registry/imr-details/women-of-reproductive-age-(15-49-years)-population-(thousands) (accessed on 23 September 2022).
- Goggins, M.; Overbeek, K.A.; Brand, R.; Syngal, S.; Del Chiaro, M.; Bartsch, D.K.; Bassi, C.; Carrato, A.; Farrell, J.; Fishman, E.K.; et al. Management of patients with increased risk for familial pancreatic cancer: Updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 2020, 69, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, F.C.; Roberts, N.J.; Childs, E.; Porter, N.; Rabe, K.G.; Borgida, A.; Ukaegbu, C.; Goggins, M.G.; Hruban, R.H.; Zogopoulos, G.; et al. Risk of Pancreatic Cancer Among Individuals with Pathogenic Variants in the ATM Gene. JAMA Oncol. 2021, 7, 1664–1668. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Audrezet, M.P.; Chen, J.M.; Le Marechal, C.; Ruszniewski, P.; Robaszkiewicz, M.; Raguenes, O.; Quere, I.; Scotet, V.; Ferec, C. Determination of the relative contribution of three genes-the cystic fibrosis transmembrane conductance regulator gene, the cationic trypsinogen gene, and the pancreatic secretory trypsin inhibitor gene-to the etiology of idiopathic chronic pancreatitis. Eur. J. Hum. Genet. 2002, 10, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, J.A.; Neoptolemos, J.P.; Feng, J.; Yan, J.; Jiang, Z.; Greenhalf, W.; McFaul, C.; Mountford, R.; Sommer, S.S. Increased risk of idiopathic chronic pancreatitis in cystic fibrosis carriers. Hum. Mutat. 2005, 26, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.D.; Talluri, J.; Wilcox, C.M.; Abberbock, J.N.; Tang, G.; Conwell, D.L.; Banks, P.A.; Cote, G.A.; Sherman, S.; Alkaade, S.; et al. Differences in Age at Onset of Symptoms, and Effects of Genetic Variants, in Patients with Early vs Late-Onset Idiopathic Chronic Pancreatitis in a North American Cohort. Clin. Gastroenterol. Hepatol. 2021, 19, 349–357. [Google Scholar] [CrossRef]
- Kashtan, C.E. Alport Syndrome: Achieving Early Diagnosis and Treatment. Am. J. Kidney Dis. 2021, 77, 272–279. [Google Scholar] [CrossRef]
- Pescucci, C.; Mari, F.; Longo, I.; Vogiatzi, P.; Caselli, R.; Scala, E.; Abaterusso, C.; Gusmano, R.; Seri, M.; Miglietti, N.; et al. Autosomal-dominant Alport syndrome: Natural history of a disease due to COL4A3 or COL4A4 gene. Kidney Int. 2004, 65, 1598–1603. [Google Scholar] [CrossRef] [Green Version]
- Uliana, V.; Sebastio, P.; Riva, M.; Carli, D.; Ruberto, C.; Bianchi, L.; Graziano, C.; Capelli, I.; Faletra, F.; Pillon, R.; et al. Deciphering the pathogenesis of the COL4-related hematuric nephritis: A genotype/phenotype study. Mol. Genet. Genom. Med. 2021, 9, e1576. [Google Scholar] [CrossRef]
- McCaffrey, T.; Guglieri, M.; Murphy, A.P.; Bushby, K.; Johnson, A.; Bourke, J.P. Cardiac involvement in female carriers of duchenne or becker muscular dystrophy. Muscle Nerve 2017, 55, 810–818. [Google Scholar] [CrossRef]
- Solheim, T.A.; Fornander, F.; Raja, A.A.; Mogelvang, R.; Poulsen, N.S.; Duno, M.; Bundgaard, H.; Vissing, J. Cardiac Involvement in Women with Pathogenic Dystrophin Gene Variants. Front. Neurol. 2021, 12, 707838. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, G.; de Souza, R.M.; Balestrino, R.; Schapira, A.H. Glucocerebrosidase Mutations in Parkinson Disease. J. Parkinson’s Dis. 2017, 7, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.; Schapira, A.H.V. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11, 1261. [Google Scholar] [CrossRef]
- Ibdah, J.A.; Bennett, M.J.; Rinaldo, P.; Zhao, Y.; Gibson, B.; Sims, H.F.; Strauss, A.W. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N. Engl. J. Med. 1999, 340, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, B.L. Sickle cell trait and sudden death--bringing it home. J. Natl. Med. Assoc. 2007, 99, 300–305. [Google Scholar] [PubMed]
- Shohat, M. Familial Mediterranean Fever. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Stoller, J.K.; Hupertz, V.; Aboussouan, L.S. Alpha-1 Antitrypsin Deficiency. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Doolan, B.J.; Onoufriadis, A.; Kantaputra, P.; McGrath, J.A. WNT10A, dermatology and dentistry. Br. J. Dermatol. 2021, 185, 1105–1111. [Google Scholar] [CrossRef]
- Kantaputra, P.; Kaewgahya, M.; Kantaputra, W. WNT10A mutations also associated with agenesis of the maxillary permanent canines, a separate entity. Am. J. Med. Genet. A 2014, 164A, 360–363. [Google Scholar] [CrossRef]
- Kantaputra, P.; Sripathomsawat, W. WNT10A and isolated hypodontia. Am. J. Med. Genet. A 2011, 155A, 1119–1122. [Google Scholar] [CrossRef]
- Arzoo, P.S.; Klar, J.; Bergendal, B.; Norderyd, J.; Dahl, N. WNT10A mutations account for (1/4) of population-based isolated oligodontia and show phenotypic correlations. Am. J. Med. Genet. A 2014, 164A, 353–359. [Google Scholar] [CrossRef]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Hutchcraft, M.L.; Zhang, S.; Lin, N.; Gottschalk, G.L.; Keck, J.W.; Belcher, E.A.; Sears, C.; Wang, C.; Liu, K.; Dietz, L.E.; et al. Real-World Evaluation of a Population Germline Genetic Screening Initiative for Family Medicine Patients. J. Pers. Med. 2022, 12, 1297. [Google Scholar] [CrossRef] [PubMed]
- Dotson, W.D.; Douglas, M.P.; Kolor, K.; Stewart, A.C.; Bowen, M.S.; Gwinn, M.; Wulf, A.; Anders, H.M.; Chang, C.Q.; Clyne, M.; et al. Prioritizing genomic applications for action by level of evidence: A horizon-scanning method. Clin. Pharmacol. Ther. 2014, 95, 394–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoury, M.J.; Dotson, W.D. From genes to public health: Are we ready for DNA-based population screening? Genet. Med. 2021, 23, 996–998. [Google Scholar] [CrossRef]
- Centers_for_Disease_Control_and_Prevention. Tier 1 Genomic Applications Toolkit for Public Health Departments. Available online: https://www.cdc.gov/genomics/implementation/toolkit/index.htm (accessed on 23 September 2022).
- Daca Alvarez, M.; Quintana, I.; Terradas, M.; Mur, P.; Balaguer, F.; Valle, L. The Inherited and Familial Component of Early-Onset Colorectal Cancer. Cells 2021, 10, 710. [Google Scholar] [CrossRef]
- Vasen, H.F.; Boland, C.R. Progress in genetic testing, classification, and identification of Lynch syndrome. JAMA 2005, 293, 2028–2030. [Google Scholar] [CrossRef]
- Zhang, L.; Bao, Y.; Riaz, M.; Tiller, J.; Liew, D.; Zhuang, X.; Amor, D.J.; Huq, A.; Petelin, L.; Nelson, M.; et al. Population genomic screening of all young adults in a health-care system: A cost-effectiveness analysis. Genet. Med. 2019, 21, 1958–1968. [Google Scholar] [CrossRef] [Green Version]
- Robertson, B.; Moyes, M.; Driscoll, A. 2021 Vermont Household Health Insurance Survey; Vermont Department of Health: Montpelier, VT, USA, 2022; p. 164. [Google Scholar]
- Zhang, L.; Walsh, M.F.; Jairam, S.; Mandelker, D.; Zhong, Y.; Kemel, Y.; Chen, Y.B.; Musheyev, D.; Zehir, A.; Jayakumaran, G.; et al. Fumarate hydratase FH c.1431_1433dupAAA (p.Lys477dup) variant is not associated with cancer including renal cell carcinoma. Hum. Mutat. 2020, 41, 103–109. [Google Scholar] [CrossRef]


| Count of Patients with | ||
|---|---|---|
| At Least 1 Variant | More Than 1 Variant or Hemizygous | Gene(s) 1 |
| 106 | 14 | HFE |
| 41 | 2 | SERPINA1 |
| 26 | 1 | CFTR |
| 24 | BTD | |
| 22 | 4 | CYP21A2 |
| 21 | GJB2 | |
| 18 | GALT | |
| 14 | F5 | |
| 13 | DHCR7 | |
| 12 | 1 | WNT10A |
| 10 | GAA | |
| 9 | SMN1 | |
| 8 | MUTYHPMM2 USH2A | |
| 6 | HBA1 PAH PKHD1 | |
| 5 | ACSF3 ATP7B CHEK2 F2 | |
| 4 | ACADM BRCA2 CDH23 EYS G6PC HOGA1 MEFV RMRP SLC22A5 SLC26A4 USH1C | |
| 3 | 1 (DNAH5) | ALDOB ATM BBS1 BBS10 BLM CACNA1S CBS CPT2 DNAH5 F11 FH GBE1 GLDC MITF PEX1 PEX7 PYGM RAPSN SLC37A4 |
| 2 | 2 (G6PD) | ABCC8 AGXT ALPL ASPA BRIP1 CAPN3 CNGB3 COL4A3 CRB1 CTNS DHDDS DSG2 FAH G6PD GNPTAB GRHPR HBA2 HBB HEXA HGD HPS3 MKS1 MYBPC3 MYO7A NEB PCDH15 PPT1 SERPINC1 SGCB SMARCAL1 SMPD1 TPP1 TYMP |
| 1 | 1 (ASS1) | ABC11 ACADVL ACAT1 AIRE ALMS1 AMT APOB ARSA ASL ASS1 ATP6V1B1 BCKDHA BRCA1 CAV3 CERKL CHRNE CLN3 CLN5 COL4A4 COL7A1 CYBA DBT DMD DNAI1 DNAI2 DSC2 DYSF ERCC6 ETFDH FANCA FANCG FKRP FKTN FLNC GALC GBA GCDH GLE1 HADHA HJV HOXB13 IDUA LAMB3 LDLR LIPA LMNA LPL LRPPRC MAN2B1 MCCC2 MCOLN1 MED17 MLC1 MMAA MMACHC MPL MPV17 MSH3 MTHFR MUT MYH7 NBN NF1 NR2E3 NTHL1 OAT PEX12 PEX6 PFKM PKP2 PMS2 POMGNT1 PRKAR1A RPGRIP1L SACS SDHD SGCG SGSH SLC12A3 SLC25A15 SLC7A7 TGM1 TMEM216 TP53 TRMU VPS13B XPA |
| 0 | 0 | ABCD1 ACAD9 ACOX1 ACTA2 ACTC1 ACTN2 ACVRL1 ADA ADAMTS2 ADGRG1 AGA AGL AGPS ALDH3A2 ALG6 APC AQP2 ARG1 ARSB ASNS ATP7A ATRX AXIN2 BAG3 BAP1 BARD1 BBS12 BBS2 BCKDHB BCS1L BMPR1A BMPR2 BSND CACNA1C CACNB2 CALM1 CALM2 CALM3 CASQ2 CAV1 CDC73 CDH1 CDK4 CDKN2A CEP290 CHM CIITA CLN6 CLN8 CLRN1 COL27A1 COL3A1 COL4A5 CPS1 CPT1A CRYAB CSRP3 CTSK CYBB CYP11B1 CYP11B2 CYP17A1 CYP19A1 CYP27A1 DCLRE1C DES DICER1 DLD DSP EDA EIF2B5 ELP1 EMD ENG EPCAM ERCC8 ESCO2 ETFA ETHE1 EVC EVC2 F9 FAM161A FANCC FBN1 FHL1 FLCN FMR1 GALK1 GAMT GDF2 GFM1 GJB1 GLA GLB1 GNE GNPTG GNS GP1BA GP9 GPD1L GREM1 HAMP HAX1 HCN4 HEXB HGSNAT HLCS HMGCL HPS1 HSD17B4 HSD3B2 HYAL1 HYLS1 IDS IL2RG IVD JUP KCNE1 KCNE2 KCNH2 KCNJ11 KCNJ2 KCNQ1 KIT LAMA2 LAMA3 LAMC2 LAMP2 LCA5 LDLRAP1 LHX3 LIFR LOXHD1 MAX MCCC1 MEN1 MESP2 MET MFSD8 MLH1 MMAB MMADHC MPI MSH2 MSH6 MTM1 MTRR MTTP MYH11 MYL2 MYL3 MYLK NAGLU NAGS NDRG1 NDUFAF5 NDUFS6 NF2 NKX2-5 NPC1 NPC2 NPHS1 NPHS2 NTRK1 OPA3 OTC PALB2 PC PCCA PCCB PCSK9 PDGFRA PDHA1 PDHB PEX10 PEX2 PHGDH PLN POLD1 POLE PRKAG2 PRKG1 PROC PROP1 PROS1 PRPS1 PSAP PTCH1 PTEN PTS PUS1 RAB23 RAD51C RAD51D RAG2 RARS2 RB1 RBM20 RDH12 RET RPE65 RS1 RTEL1 RYR1 RYR2 SAMHD1 SCN5A SDHA SDHAF2 SDHB SDHC SEPSECS SGCA SGCD SLC12A6 SLC17A5 SLC25A13 SLC26A2 SLC35A3 SLC39A4 SLC40A1 SLC4A11 SLC6A8 SMAD3 SMAD4 SMARCA4 SMARCB1 STAR STK11 SUMF1 TAT TCAP TCIRG1 TECPR2 TFR2 TGFB2 TGFB3 TGFBR1 TGFBR2 TH TMEM127 TMEM43 TNNC1 TNNI3 TNNT2 TPM1 TSC1 TSC2 TSFM TTPA VCL VHL VPS13A VPS45 VRK1 VSX2 WT1 XPC ZFYVE26 |
| Clinical Class | Class Definition | Gene-Result Count (Unique) | Patients 3 (%) | Allele Transmission Frequency | Offspring Health Risk Frequency |
|---|---|---|---|---|---|
| Negative result (Class zero) | n = 33 (11.0) | ||||
| 0 | no P/LP variants in 432 genes | n/a | 33 (11.0) | n/a | n/a |
| Class 1. Health risk | 122 (59) | n = 103 (34.3) | |||
| 1a | dominant condition, one variant, targeted disease | 51 (28) | 47 (15.7) | 0.5 | 0.5 |
| 1b | dominant condition, one variant, not targeted disease | 1 (1) | 1 (0.3) | 0.5 | 0.5 |
| 1c | recessive condition, X-linked hemizygous variant 1 | 2 (1) | 2 (0.7) | 0.5 | 0.25 |
| 1d | attenuated risk in recessive carrier (or co-dominant) | 49 (21) | 48 (16.0) | 0.5 | 0.5 |
| 1e | dominant condition, ≥2 variants in one gene | 0 (0) | 0 (0) | 0.5–1 | 0.5–1 |
| 1f | recessive condition, homo- or compound heterozygous | 17 (6) | 17 (5.7) | 1 | carrier frequency/2 |
| 1g | somatic condition (variant not germline) | 1 (1) | 1 (0.3) | 0 | 0 |
| Class 2. Carrier result, no known health risk | 508 (246) | n = 245 (81.7) | |||
| 2a | carrier, single variant | 504 (244) | 245 (81.7) | 0.5 | carrier frequency/4 |
| 2b | carrier, haplotype (two or more variants in cis) | 3 (2) | 3 (1.0) | 0.5 | carrier frequency/4 |
| 2c | double carrier, two trans variants, non-penetrant genotype | 1 (1) 2 | 1 (0.3) | 1 | carrier frequency/2 |
| Class 3. Carrier, health risk uncertain | 3 (3) | n = 3 (1.0) | |||
| 3a | ≥2 recessive variants in one gene, phase unknown | 3 (3) | 3 (1.0) | 0.5–1 | carrier frequency/4 to carrier frequency/2 |
| Total | 632 (308) | n = 300 | |||
| Gene | Increased Health Risk in Heterozygote Recessive Disease Carriers | Patient Count |
|---|---|---|
| ATM | Cancers [18,19,20] | 3 |
| CFTR1 | Chronic pancreatitis [21,22,23] | 12 |
| COL4A3, COL4A4 | Microscopic hematuria, age-related proteinuria, and chronic kidney disease [24,25,26] | 3 |
| DMD | Late-onset cardiomyopathy [27,28] | 1 |
| GBA | Parkinson disease, Lewy body dementia, or rapid eye movement sleep behavior disorders [29,30] | 1 |
| HADHA | Fatty liver of pregnancy or HELLP syndrome if carrying an affected fetus [31] | 1 |
| HBB | Vaso-occlusive events with extreme physical exertion, dehydration, and/or altitude [32] | 1 |
| MEFV | Attenuated pain syndrome [33] | 4 |
| SERPINA12 | Decreased lung function and susceptible to lung irritants [34] | 12 |
| WNT10A | Mild ectodermal dysplasia and Isolated tooth agenesis [35,36,37,38] | 11 |
| Gene | Result | Patient Count |
|---|---|---|
| CFTR | p.Phe508del and c.1210-34TG[11]T[5] (intronic) | 1 |
| HFE | p.Cys282Tyr (homozygous) | 3 |
| HFE | p.Cys282Tyr and p.His63Asp (compound heterozygous) | 4 |
| HFE | p.His63Asp (homozygous) 1 | 7 |
| SERPINA1 | p.Glu366Lys (Z allele) and p.Glu288Val (S allele) | 1 |
| WNT10A | p.Phe228Ile (homozygous) | 1 |
| Gene | Result |
|---|---|
| BRCA1 | p.Tyr1853* |
| BRCA2 | p.Gln1408* |
| BRCA2 | p.Gln2859* |
| BRCA2 | p.Glu2846Glyfs*22 |
| BRCA2 | p.Ile2588Phefs*60 |
| DSC2 | c.631-2A>G (Splice acceptor) |
| DSG2 | c.523+1G>A (Splice donor) |
| DSG2 | p.Glu1020Alafs*18 |
| HFE | p.Cys282Tyr (homozygous) |
| HFE | p.Cys282Tyr (homozygous) |
| HFE | p.Cys282Tyr (homozygous) |
| LDLR | c.2547+1G>A (Splice donor) |
| LMNA | p.Thr655Asnfs*49 |
| MYBPC3 | c.3628-41_3628-17del (Intronic) |
| MYBPC3 | p.Glu542Gln (missense and splice) |
| MYH7 | p.Lys865Arg |
| PKP2 | p.Thr50Serfs*61 |
| PMS2 | p.Lys706Serfs*19 |
| SDHD | p.Pro81Leu |
| TP53 | p.Pro151Arg (possibly mosaic) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wildin, R.S.; Gerrard, D.L.; Leonard, D.G.B. Real-World Results from Combined Screening for Monogenic Genomic Health Risks and Reproductive Risks in 300 Adults. J. Pers. Med. 2022, 12, 1962. https://doi.org/10.3390/jpm12121962
Wildin RS, Gerrard DL, Leonard DGB. Real-World Results from Combined Screening for Monogenic Genomic Health Risks and Reproductive Risks in 300 Adults. Journal of Personalized Medicine. 2022; 12(12):1962. https://doi.org/10.3390/jpm12121962
Chicago/Turabian StyleWildin, Robert S., Diana L. Gerrard, and Debra G. B. Leonard. 2022. "Real-World Results from Combined Screening for Monogenic Genomic Health Risks and Reproductive Risks in 300 Adults" Journal of Personalized Medicine 12, no. 12: 1962. https://doi.org/10.3390/jpm12121962
APA StyleWildin, R. S., Gerrard, D. L., & Leonard, D. G. B. (2022). Real-World Results from Combined Screening for Monogenic Genomic Health Risks and Reproductive Risks in 300 Adults. Journal of Personalized Medicine, 12(12), 1962. https://doi.org/10.3390/jpm12121962