
Ion Channel Involvement in Tumor Drug Resistance



Abstract
:1. Introduction
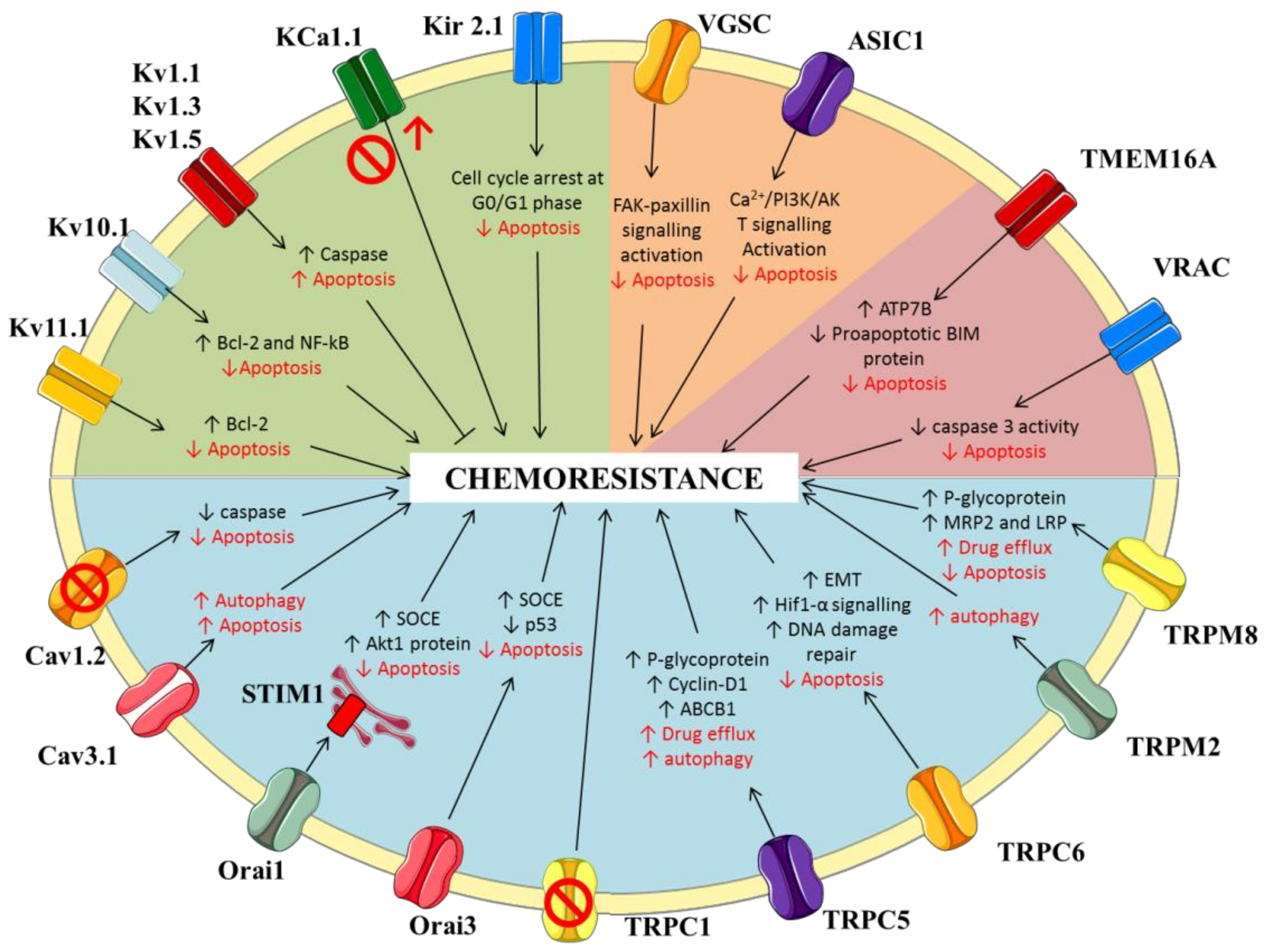
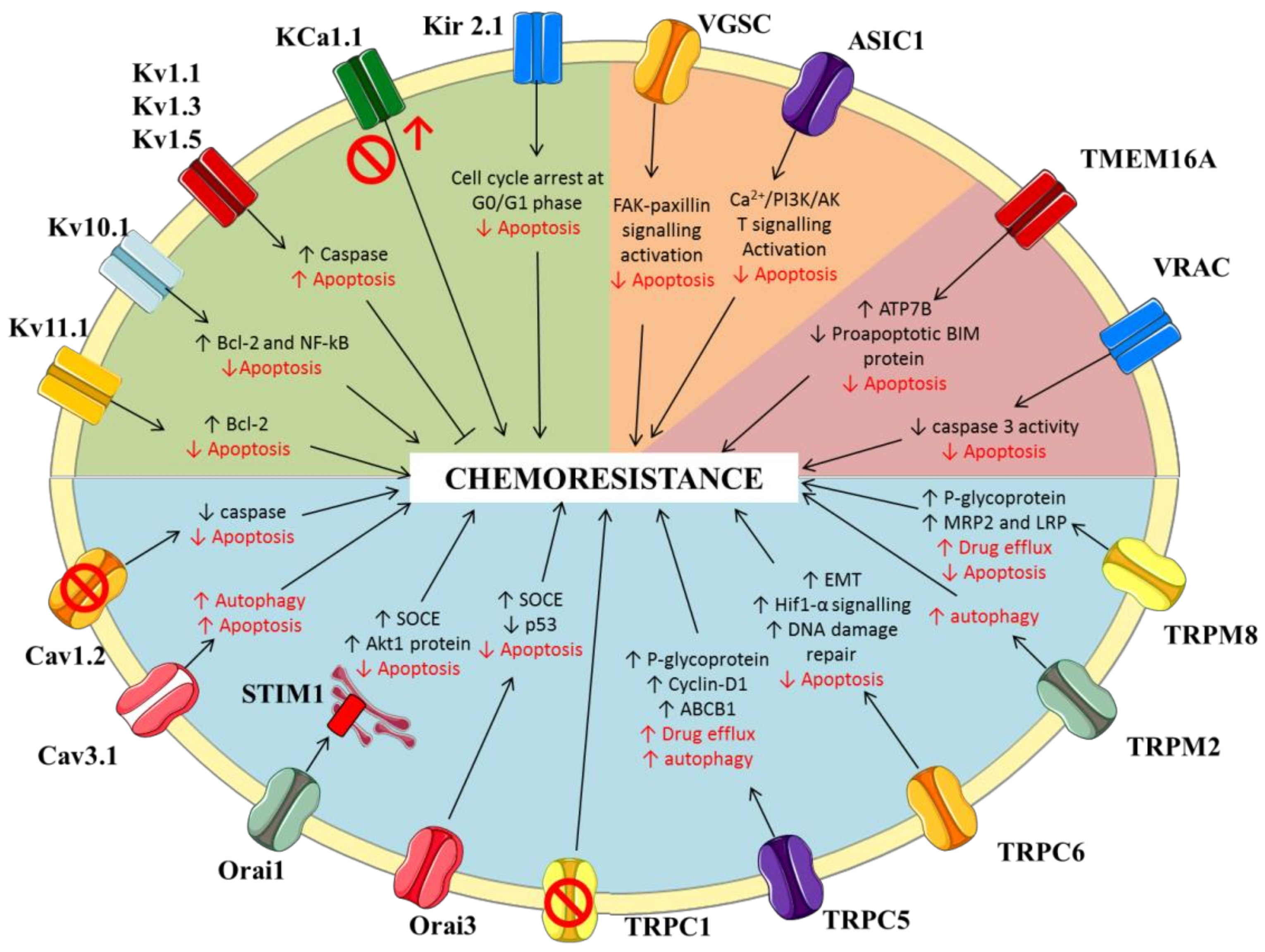
2. Potassium Channels
2.1. Voltage-Gated Potassium Channels
2.2. Calcium-Activated Potassium Channels
2.3. Inwardly-Rectifying Potassium Channels
3. Sodium Channels
3.1. Voltage-Gated Sodium Channels
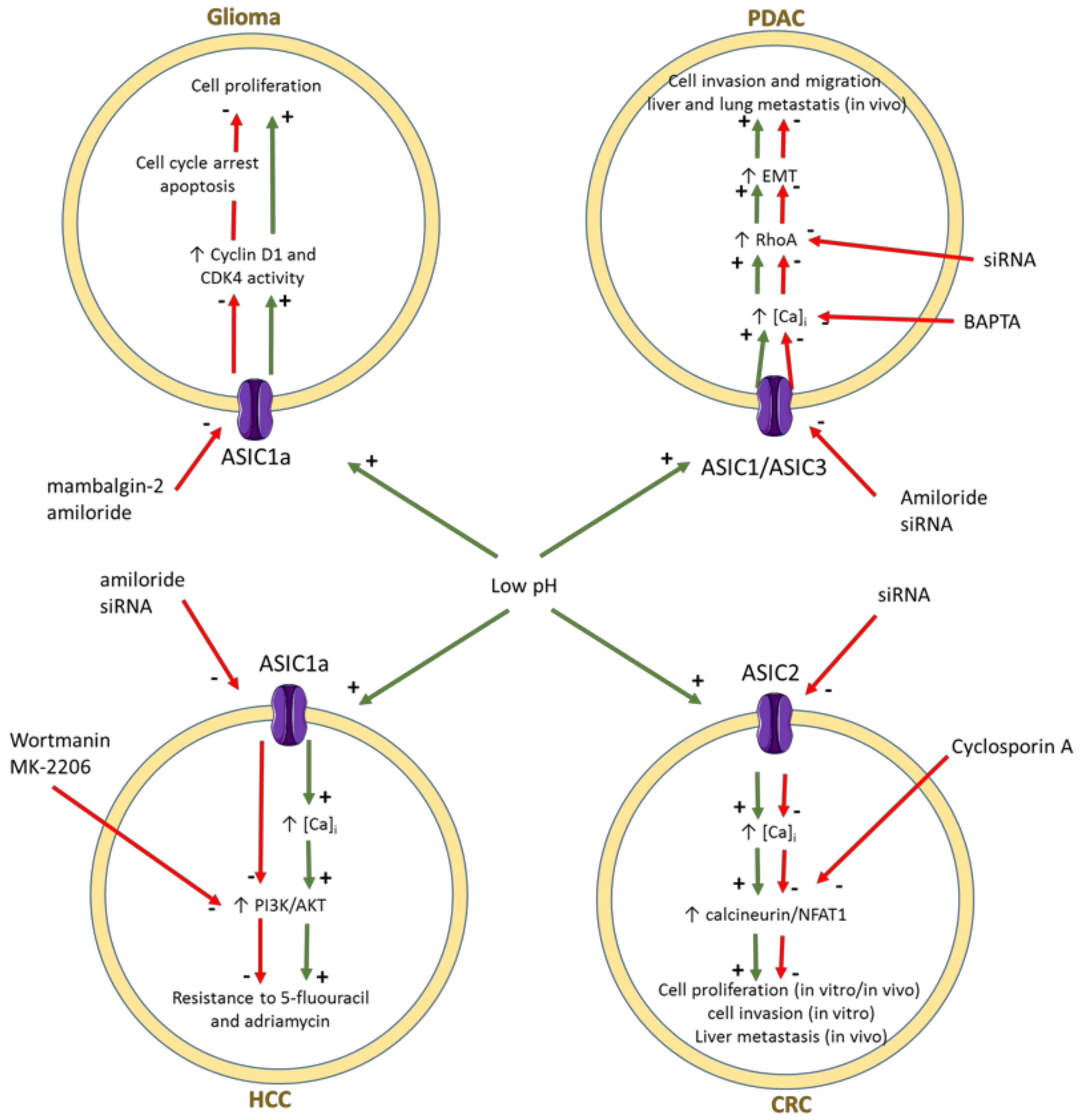
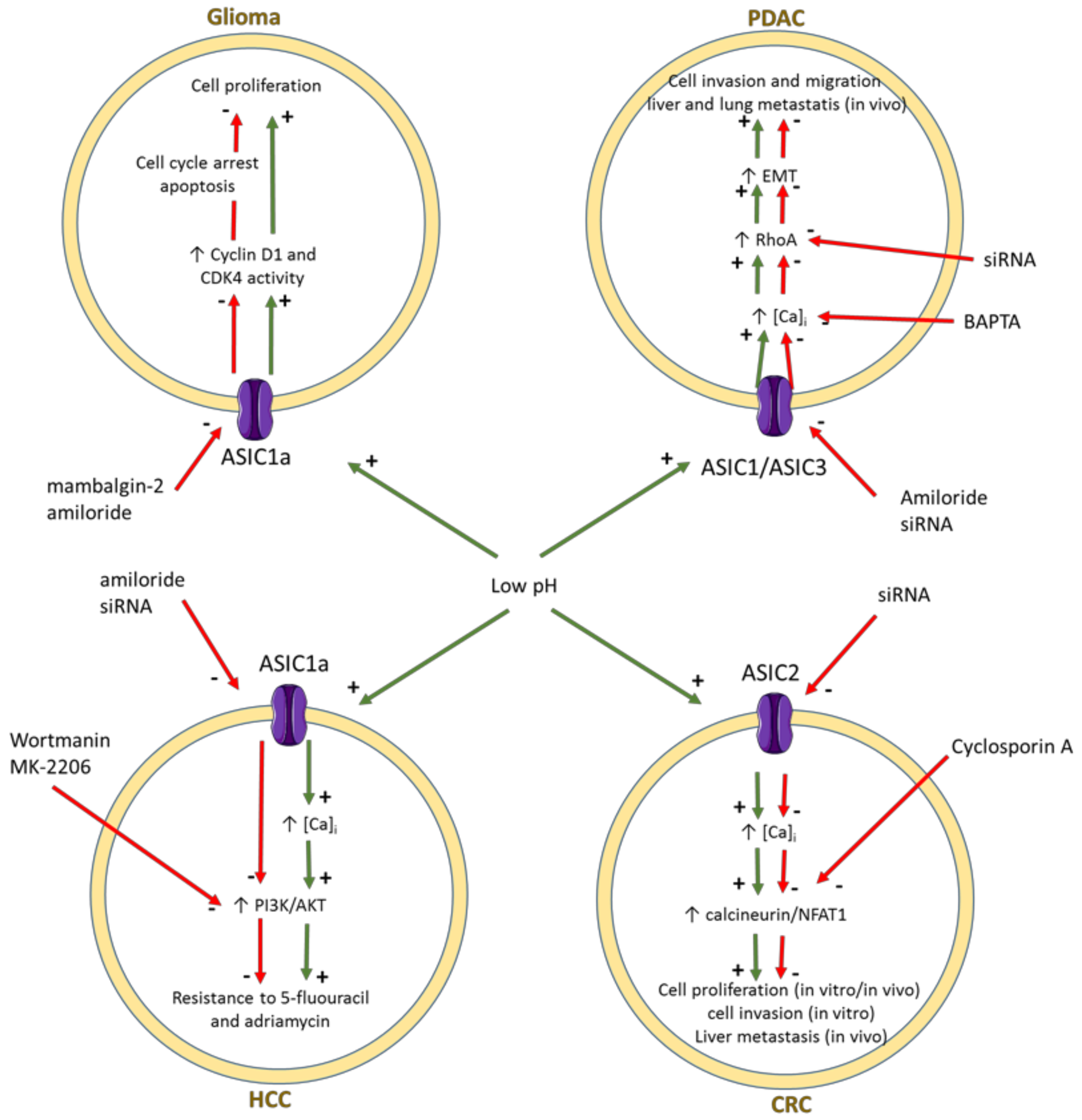
3.2. ENaC/Deg Ion Channels
4. Calcium Channels
4.1. Voltage-Gated Calcium Channels
4.2. Store-Operated Calcium Channels
4.3. Transient Receptor Potential (TRP) Channels
5. Chloride Channels
5.1. Putative Chloride Channels from the CLIC Protein Family
5.2. Calcium-Activated Chloride Channels (CaCC)
5.3. Volume Regulated Anion Channels (VRAC)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of multidrug resistance in cancer chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M. Oncochannels. Cell Calcium 2013, 53, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Djamgoz, M.B.A.; Fraser, S.P.; Brackenbury, W.J. In vivo evidence for voltage-gated sodium channel expression in carcinomas and potentiation of metastasis. Cancers 2019, 11, 1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayasu, T.; Kurisu, K.; Esquenazi, Y.; Ballester, L.Y. Ion channels and their role in the pathophysiology of gliomas. Mol. Cancer Ther. 2020, 19, 1959–1969. [Google Scholar] [CrossRef] [PubMed]
- Altamura, C.; Greco, M.R.; Carratù, M.R.; Cardone, R.A.; Desaphy, J.-F. Emerging roles for ion channels in ovarian cancer: Pathomechanisms and pharmacological treatment. Cancers 2021, 13, 668. [Google Scholar] [CrossRef]
- Hofschröer, V.; Najder, K.; Rugi, M.; Bouazzi, R.; Cozzolino, M.; Arcangeli, A.; Panyi, G.; Schwab, A. Ion channels orchestrate pancreatic ductal adenocarcinoma progression and therapy. Front. Pharmacol. 2021, 11, 586599. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, G.; Prevarskaya, N.; Schwab, A.; Lehen’kyi, V. Role of the TRP channels in pancreatic ductal adenocarcinoma development and progression. Cells 2021, 10, 1021. [Google Scholar] [CrossRef] [PubMed]
- Conti, M. Targeting ion channels for new strategies in cancer diagnosis and therapy. Curr. Clin. Pharmacol. 2007, 2, 135–144. [Google Scholar] [CrossRef]
- Felipe, A.; Bielanska, J.; Comes, N.; Vallejo, A.; Roig, S.; Ramón, Y.; Cajal, S.; Condom, E.; Hernández-Losa, J.; Ferreres, J.C. Targeting the voltage-dependent K(+) channels Kv1.3 and Kv1.5 as tumor biomarkers for cancer detection and prevention. Curr. Med. Chem. 2012, 19, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Ouadid-Ahidouch, H.; Dhennin-Duthille, I.; Gautier, M.; Sevestre, H.; Ahidouch, A. TRP channels: Diagnostic markers and therapeutic targets for breast cancer? Trends Mol. Med. 2013, 19, 117–124. [Google Scholar] [CrossRef]
- D’Amico, M.; Gasparoli, L.; Arcangeli, A. Potassium channels: Novel emerging biomarkers and targets for therapy in cancer. Recent Pat. Anticancer Drug Discov. 2013, 8, 53–65. [Google Scholar] [CrossRef]
- Frede, J.; Fraser, S.P.; Oskay-Özcelik, G.; Hong, Y.; Ioana Braicu, E.; Sehouli, J.; Gabra, H.; Djamgoz, M.B. Ovarian cancer: Ion channel and aquaporin expression as novel targets of clinical potential. Eur. J. Cancer 2013, 49, 2331–2344. [Google Scholar] [CrossRef] [PubMed]
- Pardo, L.A.; Stühmer, W. The roles of K(+) channels in cancer. Nat. Rev. Cancer 2014, 14, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Lastraioli, E.; Iorio, J.; Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta 2015, 1848, 2685–2702. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.J.; Hu, C.G.; Zhu, Z.M.; Luo, H.L. Effect of P2X7 receptor on tumorigenesis and its pharmacological properties. Biomed. Pharmacother. 2020, 125, 109844. [Google Scholar] [CrossRef]
- Cheng, W.L.; Chen, K.Y.; Lee, K.Y.; Feng, P.H.; Wu, S.M. Nicotinic-nAChR signaling mediates drug resistance in lung cancer. J. Cancer 2020, 11, 1125–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taura, J.; Kircher, D.M.; Gameiro-Ros, I.; Slesinger, P.A. Comparison of K+ channel families. Handb. Exp. Pharmacol. 2021, 267, 1–49. [Google Scholar] [PubMed]
- Arcangeli, A.; Crociani, O.; Lastraioli, E.; Masi, A.; Pillozzi, S.; Becchetti, A. Targeting ion channels in cancer: A novel frontier in antineoplastic therapy. Curr. Med. Chem. 2009, 16, 66–93. [Google Scholar] [CrossRef]
- Bachmann, M.; Li, W.; Edwards, M.J.; Ahmad, S.A.; Patel, S.; Szabo, I.; Gulbins, E. Voltage-gated potassium channels as regulators of cell death. Front. Cell Dev. Biol. 2020, 8, 611853. [Google Scholar] [CrossRef]
- Park, H.W.; Song, M.S.; Sim, H.J.; Ryu, P.D.; Lee, S.Y. The role of the voltage-gated potassium channel, Kv2.1 in prostate cancer cell migration. BMB Rep. 2021, 54, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.; Bateman, A.; O’Kelly, I. Altered expression of two-pore domain potassium (K2P) channels in cancer. PLoS ONE 2013, 8, e74589. [Google Scholar] [CrossRef] [PubMed]
- Manoli, S.; Coppola, S.; Duranti, C.; Lulli, M.; Magni, L.; Kuppalu, N.; Nielsen, N.; Schmidt, T.; Schwab, A.; Becchetti, A.; et al. The activity of Kv 11.1 potassium channel modulates F-actin organization during cell migration of pancreatic ductal adenocarcinoma cells. Cancers 2019, 11, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becchetti, A.; Crescioli, S.; Zanieri, F.; Petroni, G.; Mercatelli, R.; Coppola, S.; Gasparoli, L.; D’Amico, M.; Pillozzi, S.; Crociani, O.; et al. The conformational state of hERG1 channels determines integrin association, downstream signaling, and cancer progression. Sci. Signal. 2017, 10, eaaf3236. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.Z.; Jiang, M.; Zhen, Y.S. HERG K+ channel expression-related chemosensitivity in cancer cells and its modulation by erythromycin. Cancer Chemother. Pharmacol. 2005, 56, 212–220. [Google Scholar] [CrossRef]
- Zhang, R.; Tian, P.; Chi, Q.; Wang, J.; Wang, Y.; Sun, L.; Liu, Y.; Tian, S.; Zhang, Q. Human ether-à-go-go-related gene expression is essential for cisplatin to induce apoptosis in human gastric cancer. Oncol. Rep. 2012, 27, 433–440. [Google Scholar]
- Pillozzi, S.; Masselli, M.; De Lorenzo, E.; Accordi, B.; Cilia, E.; Crociani, O.; Amedei, A.; Veltroni, M.; D’Amico, M.; Basso, G.; et al. Chemotherapy resistance in acute lymphoblastic leukemia requires hERG1 channels and is overcome by hERG1 blockers. Blood 2011, 117, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; D’Amico, M.; Bartoli, G.; Gasparoli, L.; Petroni, G.; Crociani, O.; Marzo, T.; Guerriero, A.; Messori, L.; Severi, M.; et al. The combined activation of KCa3.1 and inhibition of Kv11.1/hERG1 currents contribute to overcome Cisplatin resistance in colorectal cancer cells. Br. J. Cancer 2018, 118, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Hui, C.; Lan, Z.; Yue-li, L.; Li-lin, H.; Li-lin, H. Knockdown of Eag1 expression by RNA interference increases chemosensitivity to cisplatin in ovarian cancer cells. Reprod. Sci. 2015, 22, 1618–1626. [Google Scholar] [CrossRef]
- Agarwal, J.R.; Griesinger, F.; Stühmer, W.; Pardo, L.A. The potassium channel Ether à go-go is a novel prognostic factor with functional relevance in acute myeloid leukemia. Mol. Cancer 2010, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Shi, Y.; Han, Z.; Sun, L.; Fan, D. Detection of potassium currents and regulation of multidrug resistance by potassium channels in human gastric cancer cells. Cell. Biol. Int. 2007, 31, 741–747. [Google Scholar] [CrossRef]
- Leanza, L.; O’Reilly, P.; Doyle, A.; Venturini, E.; Zoratti, M.; Szegezdi, E.; Szabo, I. Correlation between potassium channel expression and sensitivity to drug-induced cell death in tumor cell lines. Curr. Pharm. Des. 2014, 20, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, P.; Pink, R.C.; Caley, D.P.; Currie, J.M.; Brooks, S.A.; Carter, D.R. Over-expression of miR-31 or loss of KCNMA1 leads to increased cisplatin resistance in ovarian cancer cells. Tumour Biol. 2016, 37, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Rosa, P.; Catacuzzeno, L.; Sforna, L.; Mangino, G.; Carlomagno, S.; Mincione, G.; Petrozza, V.; Ragona, G.; Franciolini, F.; Calogero, A. BK channels blockage inhibits hypoxia-induced migration and chemoresistance to cisplatin in human glioblastoma cells. J. Cell. Physiol. 2018, 233, 6866–6877. [Google Scholar] [CrossRef]
- Liu, H.; Huang, J.; Peng, J.; Wu, X.; Zhang, Y.; Zhu, W.; Guo, L. Upregulation of the inwardly rectifying potassium channel Kir2.1 (KCNJ2) modulates multidrug resistance of small-cell lung cancer under the regulation of miR-7 and the Ras/MAPK pathway. Mol. Cancer 2015, 14, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastraioli, E.; Guasti, L.; Crociani, O.; Polvani, S.; Hofmann, G.; Witchel, H.; Bencini, L.; Calistri, M.; Messerini, L.; Scatizzi, M.; et al. herg1 gene and HERG1 protein are overexpressed in colorectal cancers and regulate cell invasion of tumor cells. Cancer Res. 2004, 64, 606–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.W.; Yang, W.B.; Gao, S.; Wang, W.; Li, Z.; Hu, W.M.; Li, J.J.; Luo, H.S. Prognostic significance of hERG1 expression in gastric cancer. Dig. Dis. Sci. 2010, 55, 1004–1010. [Google Scholar] [CrossRef]
- Menéndez, S.T.; Rodrigo, J.P.; Alvarez-Teijeiro, S.; Villaronga, M.Á.; Allonca, E.; Vallina, A.; Astudillo, A.; Barros, F.; Suárez, C.; García-Pedrero, J.M. Role of HERG1 potassium channel in both malignant transformation and disease progression in head and neck carcinomas. Mod. Pathol. 2012, 25, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Arcangeli, A. Expression and role of hERG channels in cancer cells. Novartis Found. Symp. 2005, 266, 225–234. [Google Scholar]
- He, S.; Moutaoufik, M.T.; Islam, S.; Persad, A.; Wu, A.; Aly, K.A.; Fonge, H.; Babu, M.; Cayabyab, F.S. HERG channel and cancer: A mechanistic review of carcinogenic processes and therapeutic potential. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188355. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Pointer, K.B.; Clark, P.A.; Eliceiri, K.W.; Salamat, M.S.; Robertson, G.A.; Kuo, J.S. Administration of non-torsadogenic human ether-à-go-go-related gene inhibitors is associated with better survival for high hERG-expressing glioblastoma patients. Clin. Cancer Res. 2017, 23, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duranti, C.; Iorio, J.; Lottini, T.; Lastraioli, E.; Crescioli, S.; Bagni, G.; Lulli, M.; Capitani, C.; Bouazzi, R.; Stefanini, M.; et al. Harnessing the hERG1/β1 integrin complex via a novel bispecific single-chain antibody: An effective strategy against solid cancers. Mol. Cancer Ther. 2021, 20, 1338–1349. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; Brizzi, M.F.; Balzi, M.; Crociani, O.; Cherubini, A.; Guasti, L.; Bartolozzi, B.; Becchetti, A.; Wanke, E.; Bernabei, P.A.; et al. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia 2002, 16, 1791–1798. [Google Scholar] [CrossRef]
- Yu, S.P.; Yeh, C.H.; Gottron, F.; Wang, X.; Grabb, M.C.; Choi, D.W. Role of the outward delayed rectifier K+ current in ceramide-induced caspase activation and apoptosis in cultured cortical neurons. J. Neurochem. 1999, 73, 933–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomomura, M.; Furuichi, T. Apoptosis-associated tyrosine kinase (AATYK) has differential Ca2+-dependent phosphorylation states in response to survival and apoptotic conditions in cerebellar granule cells. J. Biol. Chem. 2005, 280, 35157–35163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, N.; Yang, G.S.; Zhang, T.Y.; Chang, N.; Kang, Y.H.; Zhou, Q.; Fan, P.S. Upregulation of KCNMA1 facilitates the reversal effect of verapamil on the chemoresistance to cisplatin of esophageal squamous cell carcinoma cells. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1869–1880. [Google Scholar] [PubMed]
- Bauer, D.; Werth, F.; Nguyen, H.A.; Kiecker, F.; Eberle, J. Critical role of reactive oxygen species (ROS) for synergistic enhancement of apoptosis by vemurafenib and the potassium channel inhibitor TRAM-34 in melanoma cells. Cell Death Dis. 2017, 8, e2594. [Google Scholar] [CrossRef]
- Ohya, S.; Kajikuri, J.; Endo, K.; Kito, H.; Elboray, E.E.; Suzuki, T. Ca2+ -activated K+ channel KCa 1.1 as a therapeutic target to overcome chemoresistance in three-dimensional sarcoma spheroid models. Cancer Sci. 2021, 112, 3769–3783. [Google Scholar] [CrossRef]
- Brackenbury, W.J. Voltage-gated sodium channels and metastatic disease. Channels 2012, 6, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Fraser, S.P.; Ozerlat-Gunduz, I.; Brackenbury, W.J.; Fitzgerald, E.M.; Campbell, T.M.; Coombes, R.C.; Djamgoz, M.B. Regulation of voltage-gated sodium channel expression in cancer: Hormones, growth factors and auto-regulation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130105. [Google Scholar] [CrossRef] [Green Version]
- Angus, M.; Ruben, P. Voltage gated sodium channels in cancer and their potential mechanisms of action. Channels 2019, 13, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, S.; Rollin, J.; Barascu, A.; Besson, P.; Raynal, P.I.; Iochmann, S.; Lei, M.; Bougnoux, P.; Gruel, Y.; Le Guennec, J.Y. Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int. J. Biochem. Cell Biol. 2007, 39, 774–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, T.M.; Main, M.J.; Fitzgerald, E.M. Functional expression of the voltage-gated Na⁺-channel Nav1.7 is necessary for EGF-mediated invasion in human non-small cell lung cancer cells. J. Cell Sci. 2013, 126 Pt 21, 4939–4949. [Google Scholar] [PubMed] [Green Version]
- Roger, S.; Gillet, L.; Le Guennec, J.Y.; Besson, P. Voltage-gated sodium channels and cancer: Is excitability their primary role? Front. Pharmacol. 2015, 6, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, F.M.; Stewart, T.A.; Thompson, E.W.; Monteith, G.R. Targeting EMT in cancer: Opportunities for pharmacological intervention. Trends Pharmacol. Sci. 2014, 35, 479–488. [Google Scholar] [CrossRef]
- Eren, O.O.; Ozturk, M.A.; Sonmez, O.U.; Oyan, B. Voltage-gated sodium channel blockers can augment the efficacy of chemotherapeutics by their inhibitory effect on epithelial-mesenchymal transition. Med. Hypotheses 2015, 84, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Gradek, F.; Lopez-Charcas, O.; Chadet, S.; Poisson, L.; Ouldamer, L.; Goupille, C.; Jourdan, M.L.; Chevalier, S.; Moussata, D.; Besson, P.; et al. Sodium channel Nav1.5 controls epithelial-to-mesenchymal transition and invasiveness in breast cancer cells through its regulation by the salt-inducible kinase-1. Sci. Rep. 2019, 9, 18652. [Google Scholar] [CrossRef]
- Yamashita, N.; Hamada, H.; Tsuruo, T.; Ogata, E. Enhancement of voltage-gated Na+ channel current associated with multidrug resistance in human leukemia cells. Cancer Res. 1987, 47, 3736–3741. [Google Scholar]
- Liu, C.; Yu, M.; Li, Y.; Wang, H.; Xu, C.; Zhang, X.; Li, M.; Guo, H.; Ma, D.; Guo, X. Lidocaine inhibits the metastatic potential of ovarian cancer by blocking NaV 1.5-mediated EMT and FAK/Paxillin signaling pathway. Cancer Med. 2021, 10, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Sui, Q.; Peng, J.; Han, K.; Lin, J.; Zhang, R.; Ou, Q.; Qin, J.; Deng, Y.; Zhou, W.; Kong, L.; et al. Voltage-gated sodium channel Nav1.5 promotes tumor progression and enhances chemosensitivity to 5-fluorouracil in colorectal cancer. Cancer Lett. 2021, 500, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, S.C.; Gannon, K.P.; Stec, D.E.; Drummond, H.A. ENaC proteins contribute to VSMC migration. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H3076–H3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondarava, M.; Li, T.; Endl, E.; Wehner, F. alpha-ENaC is a functional element of the hypertonicity-induced cation channel in HepG2 cells and it mediates proliferation. Pflugers Arch. 2009, 458, 675–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ware, A.W.; Harris, J.J.; Slatter, T.L.; Cunliffe, H.E.; McDonald, F.J. The epithelial sodium channel has a role in breast cancer cell proliferation. Breast Cancer Res. Treat. 2021, 187, 31–43. [Google Scholar] [CrossRef]
- Berdiev, B.K.; Xia, J.; McLean, L.A.; Markert, J.M.; Gillespie, G.Y.; Mapstone, T.B.; Naren, A.P.; Jovov, B.; Bubien, J.K.; Ji, H.L.; et al. Acid-sensing ion channels in malignant gliomas. J. Biol. Chem. 2003, 278, 15023–15034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, N.; Bartoszewski, R.; Qadri, Y.J.; Bebok, Z.; Bubien, J.K.; Fuller, C.M.; Benos, D.J. Knockdown of ASIC1 and epithelial sodium channel subunits inhibits glioblastoma whole cell current and cell migration. J. Biol. Chem. 2009, 284, 24526–24541. [Google Scholar] [CrossRef] [Green Version]
- Bychkov, M.; Shulepko, M.; Osmakov, D.; Andreev, Y.; Sudarikova, A.; Vasileva, V.; Pavlyukov, M.S.; Latyshev, Y.A.; Potapov, A.A.; Kirpichnikov, M.; et al. Mambalgin-2 induces cell cycle arrest and apoptosis in glioma cells via interaction with ASIC1a. Cancers 2020, 12, 1837. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.H.; Song, J.W.; Li, W.; Liu, X.; Cao, L.; Wan, L.M.; Tan, Y.X.; Ji, S.P.; Liang, Y.M.; Gong, F. The acid-sensing ion channel, ASIC2, promotes invasion and metastasis of colorectal cancer under acidosis by activating the calcineurin/NFAT1 axis. J. Exp. Clin. Cancer Res. 2017, 36, 130. [Google Scholar] [CrossRef]
- Zhu, S.; Zhou, H.Y.; Deng, S.C.; Deng, S.J.; He, C.; Li, X.; Chen, J.Y.; Jin, Y.; Hu, Z.L.; Wang, F.; et al. ASIC1 and ASIC3 contribute to acidity-induced EMT of pancreatic cancer through activating Ca2+/RhoA pathway. Cell Death Dis. 2017, 8, e2806. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, T.; Wu, C.; Xia, Q.; Xu, D. ASIC1a mediates the drug resistance of human hepatocellular carcinoma via the Ca2+/PI3-kinase/AKT signaling pathway. Lab. Investig. 2017, 97, 53–69. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nature 1997, 386, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Bong, A.H.L.; Monteith, G.R. Calcium signaling and the therapeutic targeting of cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1786–1794. [Google Scholar] [CrossRef] [PubMed]
- Parys, J.B.; Bultynck, G. Ca2+ signaling and cell death: Focus on the role of Ca2+ signals in the regulation of cell death & survival processes in health, disease and therapy. Cell Calcium 2018, 70, 1–2. [Google Scholar]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Lai, M.D.; Phan, N.N.; Sun, Z.; Lin, Y.C. Meta-analysis of public microarray datasets reveals voltage-gated calcium gene signatures in clinical cancer patients. PLoS ONE 2015, 10, e0125766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.Y.; Zhang, P.P.; Zhou, W.P.; Yu, J.Y.; Yao, Z.H.; Chu, J.F.; Yao, S.N.; Wang, C.; Lone, W.; Xia, Q.X.; et al. L-type Cav 1.2 calcium channel-α-1C regulates response to rituximab in diffuse large B-cell lymphoma. Clin. Cancer Res. 2019, 25, 4168–4178. [Google Scholar] [CrossRef] [Green Version]
- Bezombes, C.; Fournié, J.J.; Laurent, G. Direct effect of rituximab in B-cell-derived lymphoid neoplasias: Mechanism, regulation, and perspectives. Mol. Cancer Res. 2011, 9, 1435–1442. [Google Scholar] [CrossRef] [Green Version]
- Janas, E.; Priest, R.; Wilde, J.I.; White, J.H.; Malhotra, R. Rituxan (anti-CD20 antibody)-induced translocation of CD20 into lipid rafts is crucial for calcium influx and apoptosis. Clin. Exp. Immunol. 2005, 139, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ayer, L.M.; Lytton, J.; Deans, J.P. Store-operated cation entry mediated by CD20 in membrane rafts. J. Biol. Chem. 2003, 278, 42427–42434. [Google Scholar] [CrossRef] [Green Version]
- Sallán, M.C.; Visa, A.; Shaikh, S.; Nàger, M.; Herreros, J.; Cantí, C. T-type Ca2+ channels: T for targetable. Cancer Res. 2018, 78, 603–609. [Google Scholar] [CrossRef] [Green Version]
- Antal, L.; Martin-Caraballo, M. T-type calcium channels in cancer. Cancers 2019, 11, 134. [Google Scholar] [CrossRef] [Green Version]
- Golden, E.B.; Cho, H.-Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schonthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y.S.; Hosoda, R.; Akiyama, Y.; Sebori, R.; Wanibuchi, M.; Mikami, T.; Sugino, T.; Suzuki, K.; Maruyama, M.; Tsukamoto, M.; et al. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J. Neurooncol. 2015, 122, 11–20. [Google Scholar] [CrossRef]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef] [Green Version]
- Visa, A.; Sallán, M.C.; Maiques, O.; Alza, L.; Talavera, E.; López-Ortega, R.; Santacana, M.; Herreros, J.; Cantí, C. T-type Cav3.1 channels mediate progression and chemotherapeutic resistance in glioblastoma. Cancer Res. 2019, 79, 1857–1868. [Google Scholar] [CrossRef] [Green Version]
- Barceló, C.; Sisó, P.; Maiques, O.; García-Mulero, S.; Sanz-Pamplona, R.; Navaridas, R.; Megino, C.; Felip, I.; Urdanibia, I.; Eritja, N.; et al. T-type calcium channels as potential therapeutic targets in vemurafenib-resistant BRAFV600E melanoma. J. Investig. Dermatol. 2020, 140, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Mertens-Walker, I.; Bolitho, C.; Baxter, R.C.; Marsh, D.J. Gonadotropin-induced ovarian cancer cell migration and proliferation require extracellular signal-regulated kinase 1/2 activation regulated by calcium and protein kinase C. Endocr. Relat. Cancer 2010, 17, 335–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.Y.; Hoover, P.J.; Mullins, P.J.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, J.; Kim, M.L.; Do Heo, W.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, R.; Lewis, R.S. Structural features of STIM and Orai underlying store-operated calcium entry. Curr. Opin. Cell Biol. 2019, 57, 90–98. [Google Scholar] [CrossRef]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Stiber, J.; Hawkins, A.; Zhang, Z.S.; Wang, S.; Burch, J.; Graham, V.; Ward, C.C.; Seth, M.; Finch, E.; Malouf, N.; et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 2008, 10, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Bisaillon, J.M.; Motiani, R.K.; Gonzalez-Cobos, J.C.; Potier, M.; Halligan, K.E.; Alzawahra, W.F.; Barroso, M.; Singer, H.A.; Jourd’heuil, D.; Trebak, M. Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. Am. J. Physiol. Cell Physiol. 2010, 298, C993–C1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiani, R.K.; Hyzinski-Garcia, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflug. Arch. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.; Liu, G.; Liu, G.; Yang, W.; Honisch, S.; Pantelakos, S.; Stournaras, C.; Höning, A.; Lang, F. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget 2014, 5, 4799–4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wei, Q.; Cheng, J.; Bian, Y.; Tian, C.; Hu, Y.; Li, H. Enhanced Stim1 expression is associated with acquired chemo-resistance of cisplatin in osteosarcoma cells. Hum. Cell 2017, 30, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Hasna, J.; Hague, F.; Rodat-Despoix, L.; Geerts, D.; Leroy, C.; Tulasne, D.; Ouadid-Ahidouch, H.; Kischel, P. Orai3 calcium channel and resistance to chemotherapy in breast cancer cells: The p53 connection. Cell Death Differ. 2018, 25, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphaël, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, M.M. TRP channels as potential drug targets. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 309–330. [Google Scholar] [CrossRef] [PubMed]
- Bon, R.S.; Beech, D.J. In pursuit of small molecule chemistry for calcium-permeable non-selective TRPC channels—Mirage or pot of gold? Br. J. Pharmacol. 2013, 170, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Elzamzamy, O.M.; Penner, R.; Hazlehurst, L.A. The role of TRPC1 in modulating cancer progression. Cells 2020, 9, 388. [Google Scholar] [CrossRef] [Green Version]
- Saldías, M.P.; Maureira, D.; Orellana-Serradell, O.; Silva, I.; Lavanderos, B.; Cruz, P.; Torres, C.; Cáceres, M.; Cerda, O. TRP channels interactome as a novel therapeutic target in breast cancer. Front. Oncol. 2021, 11, 621614. [Google Scholar] [CrossRef]
- Liu, X.; Zou, J.; Su, J.; Lu, Y.; Zhang, J.; Li, L.; Yin, F. Downregulation of transient receptor potential cation channel, subfamily C, member 1 contributes to drug resistance and high histological grade in ovarian cancer. Int. J. Oncol. 2016, 48, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Cai, Y.; He, D.; Zou, C.; Zhang, P.; Lo, C.Y.; Xu, Z.; Chan, F.L.; Yu, S.; Chen, Y.; et al. Transient receptor potential channel TRPC5 is essential for P-glycoprotein induction in drug-resistant cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, 16282–16287. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Chen, Z.; Zhu, Y.; Pan, Q.; Liu, Y.; Qi, X.; Jin, L.; Jin, J.; Ma, X.; Hua, D. Inhibition of transient receptor potential channel 5 reverses 5-Fluorouracil resistance in human colorectal cancer cells. J. Biol. Chem. 2015, 290, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Liu, X.; Li, H.; Chen, Z.; Yao, X.; Jin, J.; Ma, X. TRPC5-induced autophagy promotes drug resistance in breast carcinoma via CaMKKβ/AMPKα/mTOR pathway. Sci. Rep. 2017, 7, 3158. [Google Scholar] [CrossRef]
- Wen, L.; Liang, C.; Chen, E.; Chen, W.; Liang, F.; Zhi, X.; Wei, T.; Xue, F.; Li, G.; Yang, Q.; et al. Regulation of multi-drug Resistance in hepatocellular carcinoma cells is TRPC6/calcium dependent. Sci. Rep. 2016, 6, 23269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blake, S.D.; Tweed, C.M.; McKamey, S.G.; Koh, D.W. Transient receptor potential, melastatin-2 (TRPM2) blockade: Perspectives on potential novel clinical utility in cancer. Transl. Cancer Res. 2017, 6 (Suppl. 2), 342–347. [Google Scholar] [CrossRef]
- Bao, L.; Chen, S.J.; Conrad, K.; Keefer, K.; Abraham, T.; Lee, J.P.; Wang, J.; Zhang, X.Q.; Hirschler-Laszkiewicz, I.; Wang, H.G.; et al. Depletion of the human ion channel TRPM2 in neuroblastoma demonstrates its key role in cell survival through modulation of mitochondrial reactive oxygen species and bioenergetics. J. Biol. Chem. 2016, 291, 24449–24464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, D.W.; Powell, D.P.; Blake, S.D.; Hoffman, J.L.; Hopkins, M.M.; Feng, X. Enhanced cytotoxicity in triple-negative and estrogen receptor-positive breast adenocarcinoma cells due to inhibition of the transient receptor potential melastatin-2 channel. Oncol. Rep. 2015, 34, 1589–1598. [Google Scholar] [CrossRef] [Green Version]
- Almasi, S.; Kennedy, B.E.; El-Aghil, M.; Sterea, A.M.; Gujar, S.; Partida-Sánchez, S.; El Hiani, Y. TRPM2 channel-mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J. Biol. Chem. 2018, 293, 3637–3650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Hu, G.; Gong, Y.; Yu, Q.; He, B.; Li, W.; He, Z.; Hao, W.; He, Z.; Liu, Y. Silencing of TRPM8 inhibits aggressive tumor phenotypes and enhances gemcitabine sensitivity in pancreatic cancer. Pancreatology 2018, 18, 935–944. [Google Scholar] [CrossRef]
- Landry, D.W.; Akabas, M.H.; Redhead, C.; Edelman, A.; Cragoe, E.J., Jr.; Al-Awqati, Q. Purification and reconstitution of chloride channels from kidney and trachea. Science 1989, 244, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J.; Stein, V.; Weinreich, F.; Zdebik, A.A. Molecular structure and physiological function of chloride channels. Physiol. Rev. 2002, 82, 503–568. [Google Scholar] [CrossRef]
- Ashley, R.H. Challenging accepted ion channel biology: P64 and the CLIC family of putative intracellular anion channel proteins (review). Mol. Membr. Biol. 2003, 20, 1–11. [Google Scholar] [CrossRef]
- Peretti, M.; Angelini, M.; Savalli, N.; Florio, T.; Yuspa, S.H.; Mazzanti, M. Chloride channels in cancer: Focus on chloride intracellular channel 1 and 4 (CLIC1 AND CLIC4) proteins in tumor development and as novel therapeutic targets. Biochim. Biophys. Acta 2015, 1848, 2523–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, S.; Dal Maso, T.; Chen, J.W.; Bury, M.; Wouters, J.; Michiels, C.; Le Calve, B. Transmembrane (TMEM) protein family members: Poorly characterized even if essential for the metastatic process. Semin. Cancer Biol. 2020, 60, 96–106. [Google Scholar] [CrossRef]
- Schmit, K.; Michiels, C. TMEM proteins in cancer: A review. Front. Pharmacol. 2018, 9, 1345. [Google Scholar] [CrossRef] [Green Version]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef]
- Schroeder, B.C.; Cheng, T.; Jan, Y.N.; Jan, L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 2008, 134, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.D.; Cho, H.; Koo, J.Y.; Tak, M.H.; Cho, Y.; Shim, W.S.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.M.; et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008, 455, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Galietta, L.J. Structure and function of TMEM16 proteins (anoctamins). Physiol. Rev. 2014, 94, 419–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, T.; Sakata, A.; Nishimura, S.; Eto, K.; Nagata, S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl. Acad. Sci. USA 2015, 112, 12800–12805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Zanni, E.; Gradogna, A.; Scholz-Starke, J.; Boccaccio, A. Gain of function of TMEM16E/ANO5 scrambling activity caused by a mutation associated with gnathodiaphyseal dysplasia. Cell. Mol. Life Sci. 2018, 75, 1657–1670. [Google Scholar] [CrossRef] [Green Version]
- Paulino, C.; Kalienkova, V.; Lam, A.K.M.; Neldner, Y.; Dutzler, R. Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature 2017, 552, 421–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, S.; Feng, S.; Tien, J.; Peters, C.J.; Bulkley, D.; Lolicato, M.; Zhao, J.; Zuberbühler, K.; Ye, W.; Qi, L.; et al. Cryo-EM structures of the TMEM16A calcium-activated chloride channel. Nature 2017, 552, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Hartzell, C.; Putzier, I.; Arreola, J. Calcium-activated chloride channels. Ann. Rev. Physiol. 2005, 67, 719–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.; Duran, C.; Qu, Z.; Cui, Y.Y.; Hartzell, H.C. Explaining calcium-dependent gating of anoctamin-1 chloride channels requires a revised topology. Circ. Res. 2012, 110, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, R.; Cabrita, I.; Schreiber, R.; Kunzelmann, K. TMEM16A is indispensable for basal mucus secretion in airways and intestine. FASEB J. 2019, 33, 4502–4512. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Centeio, R.; Wanitchakool, P.; Cabrita, I.; Benedetto, R.; Saha, T.; Hoque, K.M.; Schreiber, R. Control of ion transport by Tmem16a expressed in murine intestine. Front. Physiol. 2019, 10, 1262. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, L.; Caputo, A.; Galietta, L.J. TMEM16A protein: A new identity for Ca2+-dependent Cl−channels. Physiology 2010, 25, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Leo, M.D.; Narayanan, D.; Kuruvilla, K.P.; Jaggar, J.H. Local coupling of TRPC6 to ANO1/TMEM16A channels in smooth muscle cells amplifies vasoconstriction in cerebral arteries. Am. J. Physiol. Cell Physiol. 2016, 310, C1001–C1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Gu, M.; Gao, C.; Chen, B.; Yang, J.; Xie, X.; Wang, X.; Sun, J.; Wang, J. The prognostic value and mechanisms of TMEM16A in human cancer. Front. Mol. Biosci. 2021, 8, 542156. [Google Scholar] [CrossRef]
- Crottes, D.; Jan, L.Y. The multifaceted role of TMEM16A in cancer. Cell Calcium 2019, 82, 102050. [Google Scholar] [CrossRef]
- Huang, X.; Godfrey, T.E.; Gooding, W.E.; McCarty, K.S., Jr.; Gollin, S.M. Comprehensive genome and transcriptome analysis of the 11q13 amplicon in human oral cancer and synteny to the 7F5 amplicon in murine oral carcinoma. Genes Chromosomes Cancer 2006, 45, 1058–1069. [Google Scholar] [CrossRef]
- Shiwarski, D.J.; Shao, C.; Bill, A.; Kim, J.; Xiao, D.; Bertrand, C.A.; Seethala, R.S.; Sano, D.; Myers, J.N.; Ha, P.; et al. To “grow” or “go”: TMEM16A expression as a switch between tumor growth and metastasis in SCCHN. Clin. Cancer Res. 2014, 20, 4673–4688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Liu, F.; Ji, K.; Liu, N.; He, Y.; Zhang, W.; Wang, L. MicroRNA-381 inhibits the metastasis of gastric cancer by targeting TMEM16A expression. J. Exp. Clin. Cancer Res. 2017, 36, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokutani, Y.; Uemura, M.; Munakata, K.; Okuzaki, D.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Murata, K.; Takemasa, I.; et al. Down-regulation of microRNA-132 is associated with poor prognosis of colorectal cancer. Ann. Surg. Oncol. 2016, 23, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, A.; Duvvuri, U.; Kiselyov, K. Copper-dependent ATP7B up-regulation drives the resistance of TMEM16A-overexpressing head-and-neck cancer models to platinum toxicity. Biochem. J. 2019, 476, 3705–3719. [Google Scholar] [CrossRef] [PubMed]
- Kalayda, G.V.; Wagner, C.H.; Buss, I.; Reedijk, J.; Jaehde, U. Altered localisation of the copper efflux transporters ATP7A and ATP7B associated with cisplatin resistance in human ovarian carcinoma cells. BMC Cancer 2008, 8, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, K.K.; Berkey, B.A.; Tu, X.; Zhang, H.Z.; Katz, R.; Hammond, E.H.; Fu, K.K.; Milas, L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002, 62, 7350–7356. [Google Scholar]
- Temam, S.; Kawaguchi, H.; El-Naggar, A.K.; Jelinek, J.; Tang, H.; Liu, D.D.; Lang, W.; Issa, J.P.; Lee, J.J.; Mao, L. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J. Clin. Oncol. 2007, 25, 2164–2170. [Google Scholar] [CrossRef]
- Cassell, A.; Grandis, J.R. Investigational EGFR-targeted therapy in head and neck squamous cell carcinoma. Expert Opin. Investig. Drugs 2010, 19, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bill, A.; Gutierrez, A.; Kulkarni, S.; Kemp, C.; Bonenfant, D.; Voshol, H.; Duvvuri, U.; Gaither, L.A. ANO1/TMEM16A interacts with EGFR and correlates with sensitivity to EGFR-targeting therapy in head and neck cancer. Oncotarget 2015, 6, 9173–9188. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, S.; Bill, A.; Godse, N.R.; Khan, N.I.; Kass, J.I.; Steehler, K.; Kemp, C.; Davis, K.; Bertrand, C.A.; Vyas, A.R.; et al. TMEM16A/ANO1 suppression improves response to antibody-mediated targeted therapy of EGFR and HER2/ERBB2. Genes Chromosomes Cancer 2017, 56, 460–471. [Google Scholar] [CrossRef]
- Godse, N.R.; Khan, N.; Yochum, Z.A.; Gomez-Casal, R.; Kemp, C.; Shiwarski, D.J.; Seethala, R.S.; Kulich, S.; Seshadri, M.; Burns, T.F.; et al. TMEM16A/ANO1 inhibits apoptosis via downregulation of Bim expression. Clin. Cancer Res. 2017, 23, 7324–7332. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, M.; Inoue, T.; Kito, H.; Niwa, S.; Suzuki, T.; Muraki, K.; Ohya, S. Transcriptional repression of HER2 by ANO1 Cl(-) channel inhibition in human breast cancer cells with resistance to trastuzumab. Biochem. Biophys. Res. Commun. 2017, 482, 188–194. [Google Scholar] [CrossRef]
- Cahalan, M.D.; Lewis, R.S. Role of potassium and chloride channels in volume regulation by T lymphocytes. Soc. Gen. Physiol. Ser. 1988, 43, 281–301. [Google Scholar]
- Hazama, A.; Okada, Y. Ca2+ sensitivity of volume-regulatory K+ and Cl- channels in cultured human epithelial cells. J. Physiol. 1988, 402, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 2016, 17, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.H.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. The concise guide to pharmacology 2019/20: Introduction and other protein targets. Br. J. Pharmacol. 2019, 176 (Suppl. 1), S1–S20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Okada, Y.; Nilius, B. Biophysics and physiology of the volume-regulated anion channel (VRAC)/volume-sensitive outwardly rectifying anion channel (VSOR). Pflug. Arch. 2016, 468, 371–383. [Google Scholar] [CrossRef]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of ion channels and transporters in cell migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef]
- Maeno, E.; Ishizaki, Y.; Kanaseki, T.; Hazama, A.; Okada, Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9487–9492. [Google Scholar] [CrossRef] [Green Version]
- Feustel, P.J.; Jin, Y.; Kimelberg, H.K. Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke 2004, 35, 1164–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisadome, K.; Koyama, T.; Kimura, C.; Droogmans, G.; Ito, Y.; Oike, M. Volume-regulated anion channels serve as an auto/paracrine nucleotide release pathway in aortic endothelial cells. J. Gen. Physiol. 2002, 119, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Dubin, A.E.; Mathur, J.; Tu, B.; Reddy, K.; Miraglia, L.J.; Reinhardt, J.; Orth, A.P.; Patapoutian, A. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 2014, 157, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Voss, F.K.; Ullrich, F.; Munch, J.; Lazarow, K.; Lutter, D.; Mah, N.; Andrade-Navarro, M.A.; von Kries, J.P.; Stauber, T.; Jentsch, T.J. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 2014, 344, 634–638. [Google Scholar] [CrossRef] [Green Version]
- Abascal, F.; Zardoya, R. LRRC8 proteins share a common ancestor with pannexins, and may form hexameric channels involved in cell-cell communication. Bioessays 2012, 34, 551–560. [Google Scholar] [CrossRef] [Green Version]
- Deneka, D.; Sawicka, M.; Lam, A.K.M.; Paulino, C.; Dutzler, R. Structure of a volume-regulated anion channel of the LRRC8 family. Nature 2018, 558, 254–259. [Google Scholar] [CrossRef]
- Kasuya, G.; Nakane, T.; Yokoyama, T.; Jia, Y.; Inoue, M.; Watanabe, K.; Nakamura, R.; Nishizawa, T.; Kusakizako, T.; Tsutsumi, A.; et al. Cryo-EM structures of the human volume-regulated anion channel LRRC8. Nat. Struct. Mol. Biol. 2018, 25, 797–804. [Google Scholar] [CrossRef]
- Kefauver, J.M.; Saotome, K.; Dubin, A.E.; Pallesen, J.; Cottrell, C.A.; Cahalan, S.M.; Qiu, Z.; Hong, G.; Crowley, C.S.; Whitwam, T.; et al. Structure of the human volume regulated anion channel. eLife 2018, 7, e38461. [Google Scholar] [CrossRef]
- Kern, D.M.; Oh, S.; Hite, R.K.; Brohawn, S.G. Cryo-EM structures of the DCPIB-inhibited volume-regulated anion channel LRRC8A in lipid nanodiscs. eLife 2019, 8, e42636. [Google Scholar] [CrossRef]
- Bertelli, S.; Remigante, A.; Zuccolini, P.; Barbieri, R.; Ferrera, L.; Picco, C.; Gavazzo, P.; Pusch, M. Mechanisms of activation of LRRC8 volume regulated anion channels. Cell Physiol. Biochem. 2021, 55, 41–56. [Google Scholar]
- Ullrich, F.; Reincke, S.M.; Voss, F.K.; Stauber, T.; Jentsch, T.J. Inactivation and anion selectivity of volume-regulated anion channels (VRACs) depend on C-terminal residues of the first extracellular loop. J. Biol. Chem. 2016, 291, 17040–17048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaitán-Peñas, H.; Pusch, M.; Estevez, R. Expression of LRRC8/VRAC currents in Xenopus oocytes: Advantages and caveats. Int. J. Mol. Sci. 2018, 19, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolopoulos, V.G.; Voets, T.; Declercq, P.E.; Droogmans, G.; Nilius, B. Swelling-activated efflux of taurine and other organic osmolytes in endothelial cells. Am. J. Physiol. 1997, 273, C214–C222. [Google Scholar] [CrossRef] [PubMed]
- Lutter, D.; Ullrich, F.; Lueck, J.C.; Kempa, S.; Jentsch, T.J. Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J. Cell Sci. 2017, 130, 1122–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggermont, J.; Trouet, D.; Carton, I.; Nilius, B. Cellular function and control of volume-regulated anion channels. Cell Biochem. Biophys. 2001, 35, 263–274. [Google Scholar] [CrossRef]
- Sorensen, B.H.; Thorsteinsdottir, U.A.; Lambert, I.H. Acquired cisplatin resistance in human ovarian A2780 cancer cells correlates with shift in taurine homeostasis and ability to volume regulate. Am. J. Physiol. Cell Physiol. 2014, 307, C1071–C1080. [Google Scholar] [CrossRef] [PubMed]
- Ise, T.; Shimizu, T.; Lee, E.L.; Inoue, H.; Kohno, K.; Okada, Y. Roles of volume-sensitive Cl- channel in cisplatin-induced apoptosis in human epidermoid cancer cells. J. Membr. Biol. 2005, 205, 139–145. [Google Scholar] [CrossRef]
- Lee, E.L.; Shimizu, T.; Ise, T.; Numata, T.; Kohno, K.; Okada, Y. Impaired activity of volume-sensitive Cl- channel is involved in cisplatin resistance of cancer cells. J. Cell. Physiol. 2007, 211, 513–521. [Google Scholar] [CrossRef]
- Poulsen, K.A.; Andersen, E.C.; Hansen, C.F.; Klausen, T.K.; Hougaard, C.; Lambert, I.H.; Hoffmann, E.K. Deregulation of apoptotic volume decrease and ionic movements in multidrug-resistant tumor cells: Role of chloride channels. Am. J. Physiol. Cell Physiol. 2009, 298, C14–C25. [Google Scholar] [CrossRef]
- Min, X.J.; Li, H.; Hou, S.C.; He, W.; Liu, J.; Hu, B.; Wang, J. Dysfunction of volume-sensitive chloride channels contributes to cisplatin resistance in human lung adenocarcinoma cells. Exp. Biol. Med. 2011, 236, 483–491. [Google Scholar] [CrossRef]
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. 2015, 34, 2993–3008. [Google Scholar] [CrossRef]
- Lee, C.C.; Freinkman, E.; Sabatini, D.M.; Ploegh, H.L. The protein synthesis inhibitor blasticidin s enters mammalian cells via leucine-rich repeat-containing protein 8D. J. Biol. Chem. 2014, 289, 17124–17131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gately, D.P.; Howell, S.B. Cellular accumulation of the anticancer agent cisplatin: A review. Br. J. Cancer 1993, 67, 1171–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradogna, A.; Gaitan-Penas, H.; Boccaccio, A.; Estevez, R.; Pusch, M. Cisplatin activates volume sensitive LRRC8 channel mediated currents in Xenopus oocytes. Channels 2017, 11, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Anreddy, N.; Patel, A.; Zhang, Y.K.; Wang, Y.J.; Shukla, S.; Kathawala, R.J.; Kumar, P.; Gupta, P.; Ambudkar, S.V.; Wurpel, J.N.; et al. A-803467, a tetrodotoxin-resistant sodium channel blocker, modulates ABCG2-mediated MDR in vitro and in vivo. Oncotarget 2015, 6, 39276–39291. [Google Scholar] [CrossRef] [PubMed]
- Duranti, C.; Arcangeli, A. Ion channel targeting with antibodies and antibody fragments for cancer diagnosis. Antibodies 2019, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Haustrate, A.; Hantute-Ghesquier, A.; Prevarskaya, N.; Lehen’kyi, V. Monoclonal antibodies targeting ion channels and their therapeutic potential. Front. Pharmacol. 2019, 10, 606. [Google Scholar] [CrossRef]
- Hutchings, C.J. Mini-review: Antibody therapeutics targeting G protein-coupled receptors and ion channels. Antib. Ther. 2020, 3, 257–264. [Google Scholar] [CrossRef]
- Fraser, S.P.; Onkal, R.; Theys, M.; Bosmans, F.; Djamgoz, M.B.A. Neonatal NaV 1.5: Pharmacological distinctiveness of a cancer-related voltage-gated sodium channel splice variant. Br. J. Pharmacol. 2021, 179, 473–486. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| K+ Channel | Cancer Models | Main Results | Relationship with Chemosensitivity | Ref. |
|---|---|---|---|---|
| Kv11.1 (hERG) | Various cancer cell lines (colorectal, breast, lung) | Positive correlation between level of expression and sensitivity to vincristine, camptothecin, or paclitaxel. Overexpression of Kv11.1 increased chemosensitivity. | More expression → more sensitivity | [25] |
| gastric cancer (in vitro cell lines and in vivo mouse model) | Cisplatin increased Kv11.1 expression; Silencing Kv11.1 with siRNA decreased sensitivity to cisplatin by interfering with Bcl-2-dependent apoptosis. | Less expression/activity → less sensitivity | [26] | |
| Acute lymphoblastic leukemia (cell lines and primary cell culture, and in vivo mouse model) | Kv11.1 inhibition by blockers and siRNA reduced bone marrow mesenchymal cell-induced resistance of leukemic cells to doxorubicin, prednisone, or methotrexate. | Less expression/activity → more sensitivity | [27] | |
| colorectal cancer (in vitro cell lines and in vivo mouse model) | Increased expression/activity in cisplatin-resistant cell line; Inhibition of Kv11.1 increased cisplatin uptake and ciplastin-induced apoptosis in vitro, and overcome cisplatin resistance in vivo. | Less expression/activity → more sensitivity | [28] | |
| Kv10.1 (hEag1) | Ovarian cancer (OC) (patient biopsies and cell lines) | Overall survival longer in cisplatin-treated OC patients with lower Kv10.1 expression; Silencing of Kv10.1 increased sensitivity to cisplatin by interfering with NFkB/Bcl-2 dependent apoptosis. | Less expression/activity → more sensitivity | [29] |
| Hematological malignancies (patient biopsies, primary cells and cell lines) | Increased expression in acute myeloid leukemia patients predictive of a poor outcome; Kv10.1 inhibition by blockers or siRNA reduced cell proliferation and increased sensitivity to etoposide, cytarabine, or doxorubicin by promoting caspase activity. | Less expression/activity → more sensitivity | [30] | |
| Kv1.5 | Gastric cancer (cell lines) | Kv1.5 inhibition by K+ channel blocker or siRNA enhanced resistance to doxurubicin, 5-fluouracil, vincristine, or cisplatin, while Kv1.5 overexpression increased chemosensitivity. | Less expression/activity → less sensitivity More expression/activity → more sensitivity | [31] |
| Kv1.1, Kv1.3 | Cancer cell line panel | Expression positively correlated with cisplatin-induced cell death. | More expression/activity → more sensitivity | [32] |
| KCa1.1 (BK) | Ovarian cancer (cell lines and primary cells) | KCa1.1 expression is inversely correlated with resistance to cisplatin; Channel knockdown by siRNA increased resistance to cisplatin. | Less expression/activity → less sensitivity | [33] |
| Glioblastoma (cell line) | KCa1.1 promotes hypoxia-induced cell migration and resistance to cisplatin; KCa1.1 inhibition by paxilline increased sensitivity to cisplatin. | More expression/activity → less sensitivity Less expression/activity → more sensitivity | [34] | |
| Kir2.1 | Small cell lung cancer (patients, cell lines, and in vivo mouse model) | Increased Kir2.1 expression in patients’ cancer cells correlated with clinical stage progression and chemoresistance; Overexpression of Kir2.1 increased resistance to etoposide or cisplatin, whereas knockdown with siRNA increased chemosensitivity. | More expression/activity → less sensitivity Less expression/activity → more sensitivity | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altamura, C.; Gavazzo, P.; Pusch, M.; Desaphy, J.-F. Ion Channel Involvement in Tumor Drug Resistance. J. Pers. Med. 2022, 12, 210. https://doi.org/10.3390/jpm12020210
Altamura C, Gavazzo P, Pusch M, Desaphy J-F. Ion Channel Involvement in Tumor Drug Resistance. Journal of Personalized Medicine. 2022; 12(2):210. https://doi.org/10.3390/jpm12020210
Chicago/Turabian StyleAltamura, Concetta, Paola Gavazzo, Michael Pusch, and Jean-François Desaphy. 2022. "Ion Channel Involvement in Tumor Drug Resistance" Journal of Personalized Medicine 12, no. 2: 210. https://doi.org/10.3390/jpm12020210
APA StyleAltamura, C., Gavazzo, P., Pusch, M., & Desaphy, J.-F. (2022). Ion Channel Involvement in Tumor Drug Resistance. Journal of Personalized Medicine, 12(2), 210. https://doi.org/10.3390/jpm12020210