
Endothelial Senescence and the Chronic Vascular Diseases: Challenges and Therapeutic Opportunities in Atherosclerosis

 ,
,  , , , and
, , , and 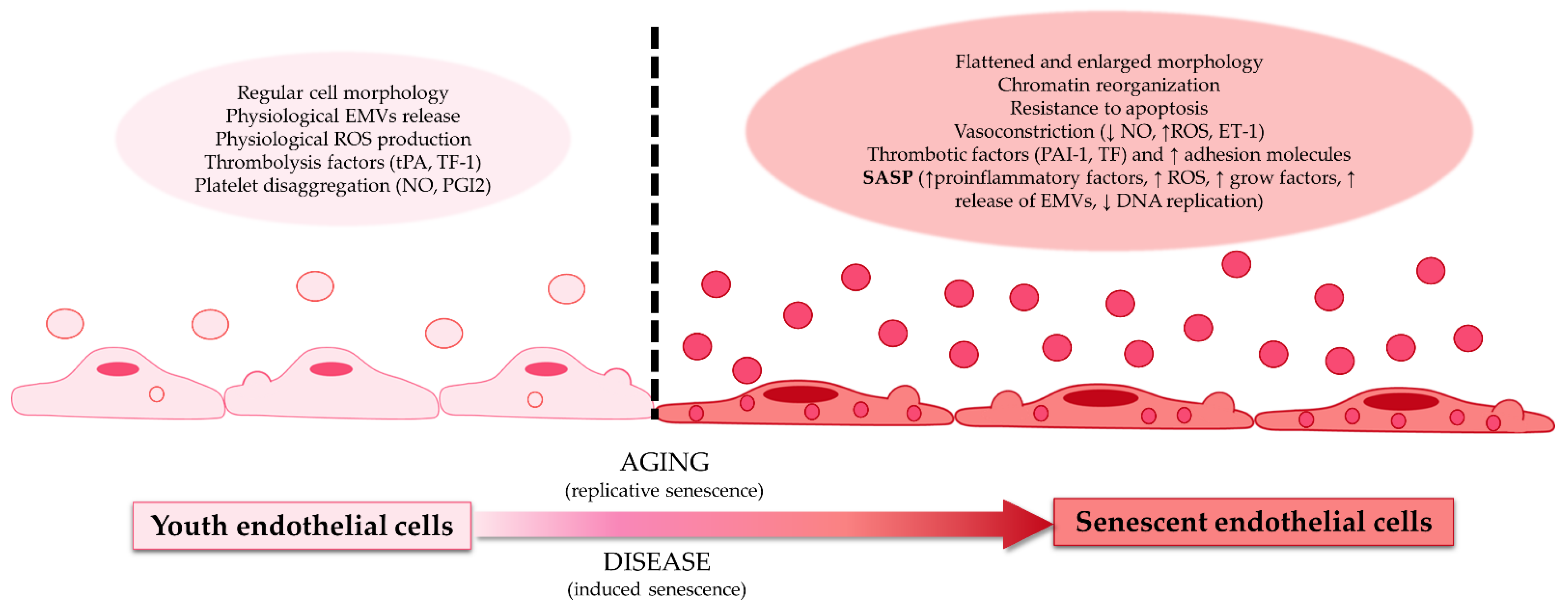{kind=link}
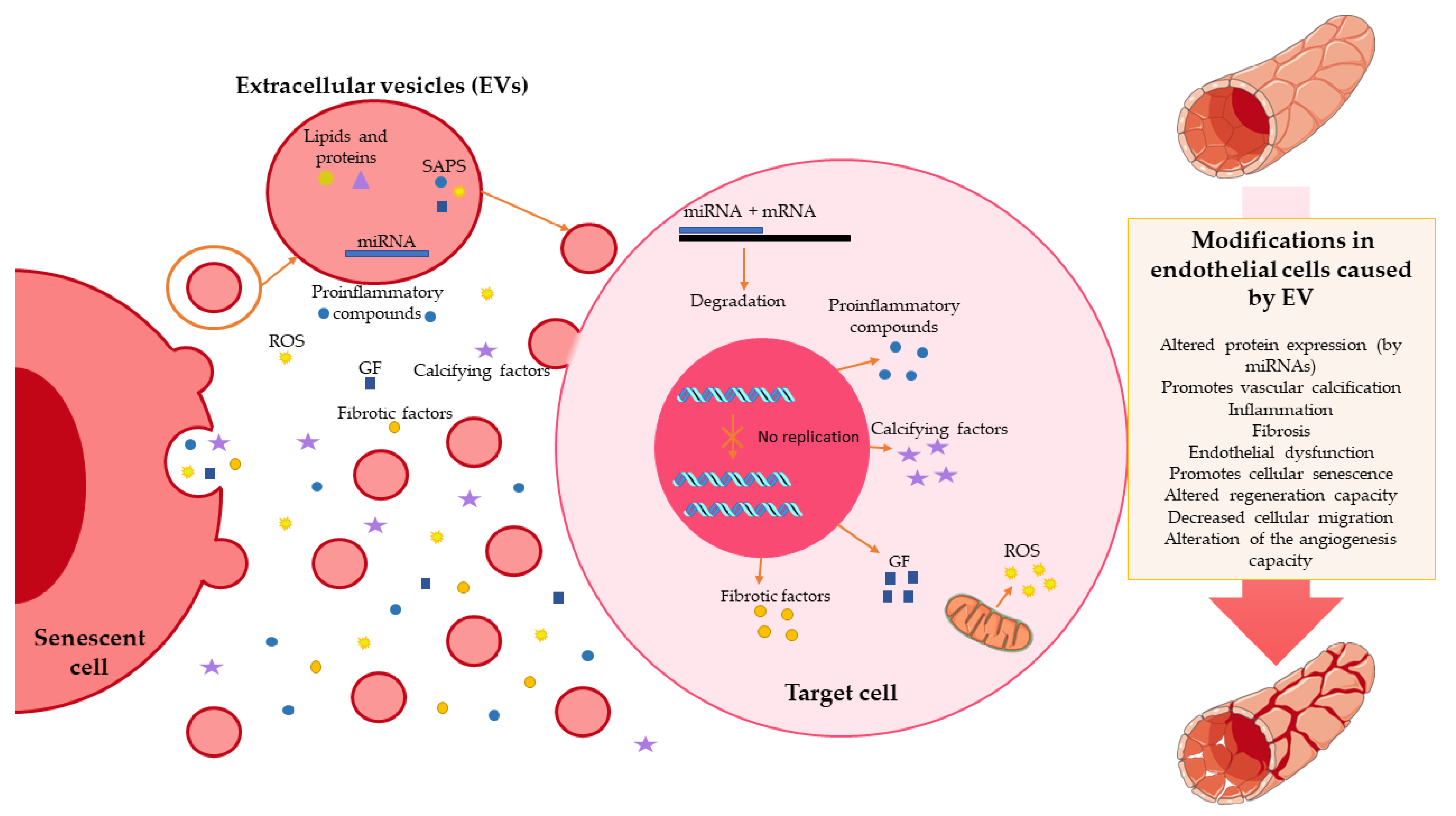{kind=link}
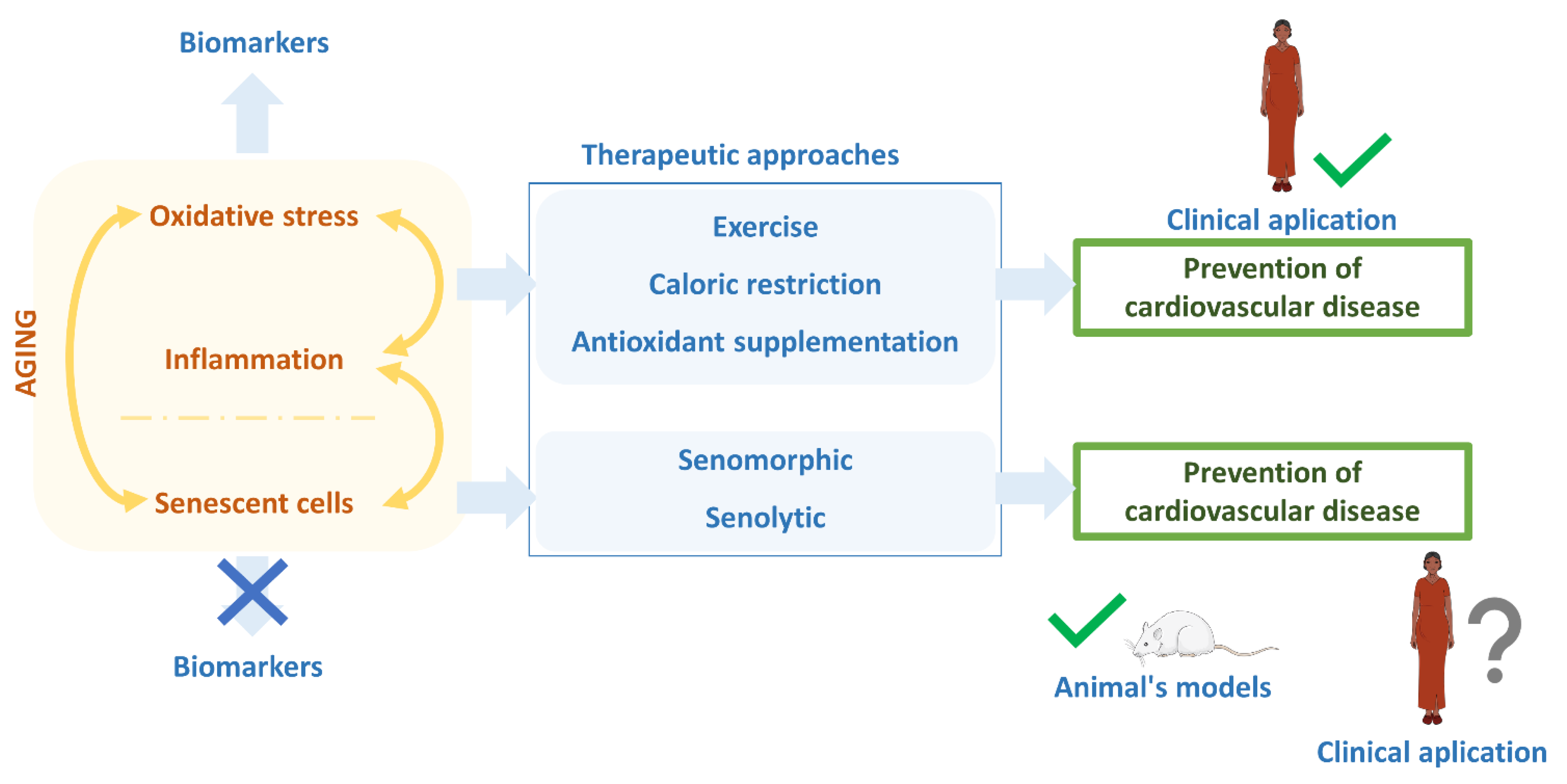{kind=link}
Abstract
:1. Introduction
2. Cellular Senescence
2.1. Factors That Induce Endothelial Senescence
2.2. Low Degree Chronic Systemic Inflammation as a Key Risk Factor of Endothelial Senescence
3. Extracellular Vesicles and Endothelial Senescence
4. Target Endothelial Senescence: A Promising Therapeutic Opportunity
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Figuer, A.; Bodega, G.; Tato, P.; Valera, G.; Serroukh, N.; Ceprian, N.; de Sequera, P.; Morales, E.; Carracedo, J.; Ramirez, R.; et al. Premature Aging in Chronic Kidney Disease: The Outcome of Persistent Inflammation beyond the Bounds. Int. J. Environ. Res. Public Health 2021, 18, 8044. [Google Scholar] [CrossRef]
- Marengoni, A.; Fratiglioni, L. Disease clusters in older adults: Rationale and need for investigation. J. Am. Geriatr. Soc 2011, 59, 2395–2396. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Recent advances in the discovery of senolytics. Mech. Ageing Dev. 2021, 200, 111587. [Google Scholar] [CrossRef]
- Shlisky, J.; Bloom, D.E.; Beaudreault, A.R.; Tucker, K.L.; Keller, H.H.; Freund-Levi, Y.; Fielding, R.A.; Cheng, F.W.; Jensen, G.L.; Wu, D.; et al. Nutritional Considerations for Healthy Aging and Reduction in Age-Related Chronic Disease. Adv. Nutr. 2017, 8, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Yazdanyar, A.; Newman, A.B. The Burden of Cardiovascular Disease in the Elderly: Morbidity, Mortality, and Costs. Clin. Geriatr. Med. 2009, 25, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.M.; Zheng, L.; Wang, Q.; Hu, Y.W. The emerging role of cell senescence in atherosclerosis. Clin. Chem. Lab. Med. 2020, 59, 27–38. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Ebert, B.L. Clonal Hematopoiesis and Atherosclerosis. N. Engl. J. Med. 2017, 377, 1401–1402. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Farr, J.N. Cellular senescence in age-related disorders. Transl. Res. 2020, 226, 96–104. [Google Scholar] [CrossRef]
- Kaplan, H.; Thompson, R.C.; Trumble, B.C.; Wann, L.S.; Allam, A.H.; Beheim, B.; Frohlich, B.; Sutherland, M.L.; Sutherland, J.D.; Stieglitz, J.; et al. Coronary atherosclerosis in indigenous South American Tsimane: A cross-sectional cohort study. Lancet 2017, 389, 1730–1739. [Google Scholar] [CrossRef]
- del Campo, L.; Hamczyk, M.R.; Andrés, V.; Martínez-González, J.; Rodríguez, C. Mecanismos de envejecimiento vascular: ¿Qué podemos aprender del síndrome de progeria de Hutchinson-Gilford? Clínica e Investigación en Arteriosclerosis 2018, 30, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Lam, E.W.; Tchkonia, T.; Kirkland, J.L.; Sun, Y. Senescent Cells: Emerging Targets for Human Aging and Age-Related Diseases. Trends Biochem. Sci. 2020, 45, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Kroemer, G. Hallmarks of Health. Cell 2021, 184, 33–63. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Feinberg, M.W. Vascular Endothelial Senescence: Pathobiological Insights, Emerging Long Noncoding RNA Targets, Challenges and Therapeutic Opportunities. Front. Physiol. 2021, 12, 693067. [Google Scholar] [CrossRef]
- Martinez de Toda, I.; Ceprian, N.; Diaz-Del Cerro, E.; De la Fuente, M. The Role of Immune Cells in Oxi-Inflamm-Aging. Cells 2021, 10, 2974. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, J.; Ramírez-Carracedo, R.; Alique, M.; Ramírez-Chamond, R. Endothelial Cell Senescence in the Pathogenesis of Endothelial Dysfunction; InTech: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Hohensinner, P.J.; Kaun, C.; Buchberger, E.; Ebenbauer, B.; Demyanets, S.; Huk, I.; Eppel, W.; Maurer, G.; Huber, K.; Wojta, J. Age intrinsic loss of telomere protection via TRF1 reduction in endothelial cells. Biochim. Biophys. Acta 2016, 1863, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Parimon, T.; Hohmann, M.S.; Yao, C. Cellular Senescence: Pathogenic Mechanisms in Lung Fibrosis. Int. J. Mol. Sci. 2021, 22, 6214. [Google Scholar] [CrossRef]
- Fenton, M.; Barker, S.; Kurz, D.J.; Erusalimsky, J.D. Cellular senescence after single and repeated balloon catheter denudations of rabbit carotid arteries. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 220–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Brodsky, S.V.; Goligorsky, D.M.; Hampel, D.J.; Li, H.; Gross, S.S.; Goligorsky, M.S. Glycated collagen I induces premature senescence-like phenotypic changes in endothelial cells. Circ. Res. 2002, 90, 1290–1298. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, S.V.; Gealekman, O.; Chen, J.; Zhang, F.; Togashi, N.; Crabtree, M.; Gross, S.S.; Nasjletti, A.; Goligorsky, M.S. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circ. Res. 2004, 94, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasile, E.; Tomita, Y.; Brown, L.F.; Kocher, O.; Dvorak, H.F. Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: Evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J. 2001, 15, 458–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [Green Version]
- Villaret, A.; Galitzky, J.; Decaunes, P.; Esteve, D.; Marques, M.A.; Sengenes, C.; Chiotasso, P.; Tchkonia, T.; Lafontan, M.; Kirkland, J.L.; et al. Adipose tissue endothelial cells from obese human subjects: Differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes 2010, 59, 2755–2763. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Liu, Y.; Huang, L.; Tan, H. Advances in epigenetic regulation of vascular aging. Rev. Cardiovasc. Med. 2019, 20, 19–25. [Google Scholar] [CrossRef]
- Durik, M.; Kavousi, M.; van der Pluijm, I.; Isaacs, A.; Cheng, C.; Verdonk, K.; Loot, A.E.; Oeseburg, H.; Bhaggoe, U.M.; Leijten, F.; et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation 2012, 126, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Uryga, A.; Gray, K.; Bennett, M. DNA Damage and Repair in Vascular Disease. Annu. Rev. Physiol. 2016, 78, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.V.; Wiley, C.D.; Velarde, M.C. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell 2015, 14, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirsch, J.; Schneider, H.; Pagel, J.I.; Rehberg, M.; Singer, M.; Hellfritsch, J.; Chillo, O.; Schubert, K.M.; Qiu, J.; Pogoda, K.; et al. Endothelial Dysfunction, and A Prothrombotic, Proinflammatory Phenotype Is Caused by Loss of Mitochondrial Thioredoxin Reductase in Endothelium. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1891–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, D.J.; Decary, S.; Hong, Y.; Trivier, E.; Akhmedov, A.; Erusalimsky, J.D. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J. Cell. Sci. 2004, 117, 2417–2426. [Google Scholar] [CrossRef] [Green Version]
- Ogami, M.; Ikura, Y.; Ohsawa, M.; Matsuo, T.; Kayo, S.; Yoshimi, N.; Hai, E.; Shirai, N.; Ehara, S.; Komatsu, R.; et al. Telomere shortening in human coronary artery diseases. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 546–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Dai, L.; Qureshi, A.R.; Witasp, A.; Lindholm, B.; Stenvinkel, P. Early Vascular Ageing and Cellular Senescence in Chronic Kidney Disease. Comput. Struct. Biotechnol. J. 2019, 17, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Invest. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Liu, H.; Ha, Y.; Tilton, R.G.; Zhang, W. Oxidative stress induces endothelial cell senescence via downregulation of Sirt6. Biomed. Res. Int. 2014, 2014, 902842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Zhang, H.; Lei, F.; Wang, R.; Lin, T.; Liao, L. Epigenetic therapy attenuates oxidative stress in BMSCs during ageing. J. Cell. Mol. Med. 2021, 26, 375–384. [Google Scholar] [CrossRef]
- El Hadri, K.; Smith, R.; Duplus, E.; El Amri, C. Inflammation, Oxidative Stress, Senescence in Atherosclerosis: Thioredoxine-1 as an Emerging Therapeutic Target. Int. J. Mol. Sci. 2021, 23, 77. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Baylis, D.; Bartlett, D.B.; Patel, H.P.; Roberts, H.C. Understanding how we age: Insights into inflammaging. Longev. Healthspan 2013, 2, 8. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Goldstein, D.R. Ageing and atherosclerosis: Vascular intrinsic and extrinsic factors and potential role of IL-6. Nat. Rev. Cardiol. 2021, 18, 58–68. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Merino, A.; Buendia, P.; Martin-Malo, A.; Aljama, P.; Ramirez, R.; Carracedo, J. Senescent CD14+CD16+ monocytes exhibit proinflammatory and proatherosclerotic activity. J. Immunol. 2011, 186, 1809–1815. [Google Scholar] [CrossRef] [Green Version]
- Cook, E.K.; Luo, M.; Rauh, M.J. Clonal hematopoiesis and inflammation: Partners in leukemogenesis and comorbidity. Exp. Hematol. 2020, 83, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Matteini, F.; Mulaw, M.A.; Florian, M.C. Aging of the Hematopoietic Stem Cell Niche: New Tools to Answer an Old Question. Front. Immunol. 2021, 12, 738204. [Google Scholar] [CrossRef]
- Caiado, F.; Pietras, E.M.; Manz, M.G. Inflammation as a regulator of hematopoietic stem cell function in disease, aging, and clonal selection. J. Exp. Med. 2021, 218, e20201541. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Yang, J.; Cao, W.; Sun, Y. Differential diagnosis of acute rejection and chronic cyclosporine nephropathy after rat renal transplantation by detection of endothelial microparticles (EMP). Med. Hypotheses. 2010, 75, 666–668. [Google Scholar] [CrossRef]
- Lee, T.H.; D’Asti, E.; Magnus, N.; Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles as mediators of intercellular communication in cancer--the emerging science of cellular ‘debris’. Semin. Immunopathol. 2011, 33, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Mezentsev, A.; Merks, R.M.; O’Riordan, E.; Chen, J.; Mendelev, N.; Goligorsky, M.S.; Brodsky, S.V. Endothelial microparticles affect angiogenesis in vitro: Role of oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1106–H1114. [Google Scholar] [CrossRef]
- Carracedo, J.; Alique, M.; Vida, C.; Bodega, G.; Ceprian, N.; Morales, E.; Praga, M.; de Sequera, P.; Ramirez, R. Mechanisms of Cardiovascular Disorders in Patients with Chronic Kidney Disease: A Process Related to Accelerated Senescence. Front. Cell. Dev. Biol. 2020, 8, 185. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.T.; Liu, B.C. Extracellular Vesicles: Opportunities and Challenges for the Treatment of Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 693–709. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008, 68, 7864–7871. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Nishikawa, M.; Takakura, Y. In Vivo Tracking of Extracellular Vesicles in Mice Using Fusion Protein Comprising Lactadherin and Gaussia Luciferase. Methods Mol. Biol. 2017, 1660, 245–254. [Google Scholar] [CrossRef]
- Alique, M.; Ramirez-Carracedo, R.; Bodega, G.; Carracedo, J.; Ramirez, R. Senescent Microvesicles: A Novel Advance in Molecular Mechanisms of Atherosclerotic Calcification. Int. J. Mol. Sci. 2018, 19, 2003. [Google Scholar] [CrossRef] [Green Version]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles Derived from Indoxyl Sulfate Treated Endothelial Cells Induce Endothelial Progenitor Cells Dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef]
- Georgatzakou, H.T.; Pavlou, E.G.; Papageorgiou, E.G.; Papassideri, I.S.; Kriebardis, A.G.; Antonelou, M.H. The Multi-Faced Extracellular Vesicles in the Plasma of Chronic Kidney Disease Patients. Front. Cell. Dev. Biol. 2020, 8, 227. [Google Scholar] [CrossRef]
- Alique, M.; Bodega, G.; Corchete, E.; Garcia-Menendez, E.; de Sequera, P.; Luque, R.; Rodriguez-Padron, D.; Marques, M.; Portoles, J.; Carracedo, J.; et al. Microvesicles from indoxyl sulfate-treated endothelial cells induce vascular calcification in vitro. Comput. Struct. Biotechnol. J. 2020, 18, 953–966. [Google Scholar] [CrossRef]
- Guerrero, F.; Carmona, A.; Obrero, T.; Jimenez, M.J.; Soriano, S.; Moreno, J.A.; Martin-Malo, A.; Aljama, P. Role of endothelial microvesicles released by p-cresol on endothelial dysfunction. Sci. Rep. 2020, 10, 10657. [Google Scholar] [CrossRef]
- Terentes-Printzios, D.; Vlachopoulos, C.; Xaplanteris, P.; Ioakeimidis, N.; Aznaouridis, K.; Baou, K.; Kardara, D.; Georgiopoulos, G.; Georgakopoulos, C.; Tousoulis, D. Cardiovascular Risk Factors Accelerate Progression of Vascular Aging in the General Population: Results from the CRAVE Study (Cardiovascular Risk Factors Affecting Vascular Age). Hypertension 2017, 70, 1057–1064. [Google Scholar] [CrossRef]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Sabath, N.; Levy-Adam, F.; Younis, A.; Rozales, K.; Meller, A.; Hadar, S.; Soueid-Baumgarten, S.; Shalgi, R. Cellular proteostasis decline in human senescence. Proc. Natl. Acad. Sci. USA 2020, 117, 31902–31913. [Google Scholar] [CrossRef] [PubMed]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for Targeting Senescent Cells in Human Disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.; Ojcius, D.M.; Wu, C.Y.; Peng, H.H.; Voisin, L.; Perfettini, J.L.; Ko, Y.F.; Young, J.D. Emerging use of senolytics and senomorphics against aging and chronic diseases. Med. Res. Rev. 2020, 40, 2114–2131. [Google Scholar] [CrossRef]
- Lagoumtzi, S.M.; Chondrogianni, N. Senolytics and senomorphics: Natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free Radic. Biol. Med. 2021, 171, 169–190. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking aging to chronic disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef] [Green Version]
- Niedernhofer, L.J.; Robbins, P.D. Senotherapeutics for healthy ageing. Nat. Rev. Drug. Discov. 2018, 17, 377. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Wang, E. Senescent Human Fibroblasts Resist Programmed Cell Death, and Failure to Suppress bcl2 Is Involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar]
- Blagosklonny, M.V. Anti-aging: Senolytics or gerostatics (unconventional view). Oncotarget 2021, 12, 1821–1835. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.M.; Kaistha, A.; Uryga, A.K.; Oc, S.; Foote, K.; Shah, A.; Finigan, A.; Figg, N.; Dobnikar, L.; Jorgensen, H.; et al. Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc. Res. 2021, 1–15. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez, R.; Ceprian, N.; Figuer, A.; Valera, G.; Bodega, G.; Alique, M.; Carracedo, J. Endothelial Senescence and the Chronic Vascular Diseases: Challenges and Therapeutic Opportunities in Atherosclerosis. J. Pers. Med. 2022, 12, 215. https://doi.org/10.3390/jpm12020215
Ramírez R, Ceprian N, Figuer A, Valera G, Bodega G, Alique M, Carracedo J. Endothelial Senescence and the Chronic Vascular Diseases: Challenges and Therapeutic Opportunities in Atherosclerosis. Journal of Personalized Medicine. 2022; 12(2):215. https://doi.org/10.3390/jpm12020215
Chicago/Turabian StyleRamírez, Rafael, Noemi Ceprian, Andrea Figuer, Gemma Valera, Guillermo Bodega, Matilde Alique, and Julia Carracedo. 2022. "Endothelial Senescence and the Chronic Vascular Diseases: Challenges and Therapeutic Opportunities in Atherosclerosis" Journal of Personalized Medicine 12, no. 2: 215. https://doi.org/10.3390/jpm12020215