
A State-of-the-Art Roadmap for Biomarker-Driven Drug Development in the Era of Personalized Therapies

 , and
, and
Abstract
:1. Introduction
2. Background
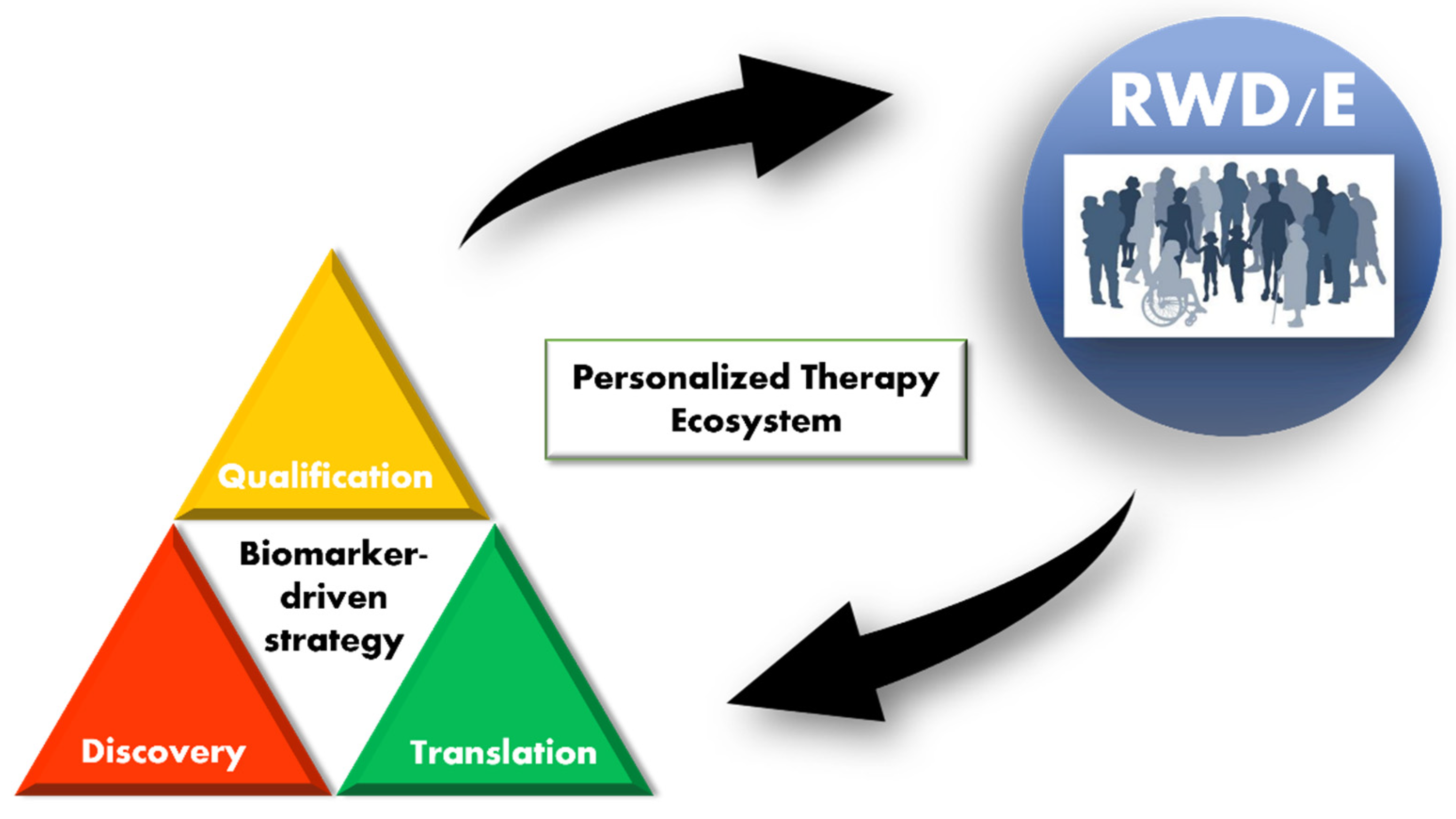
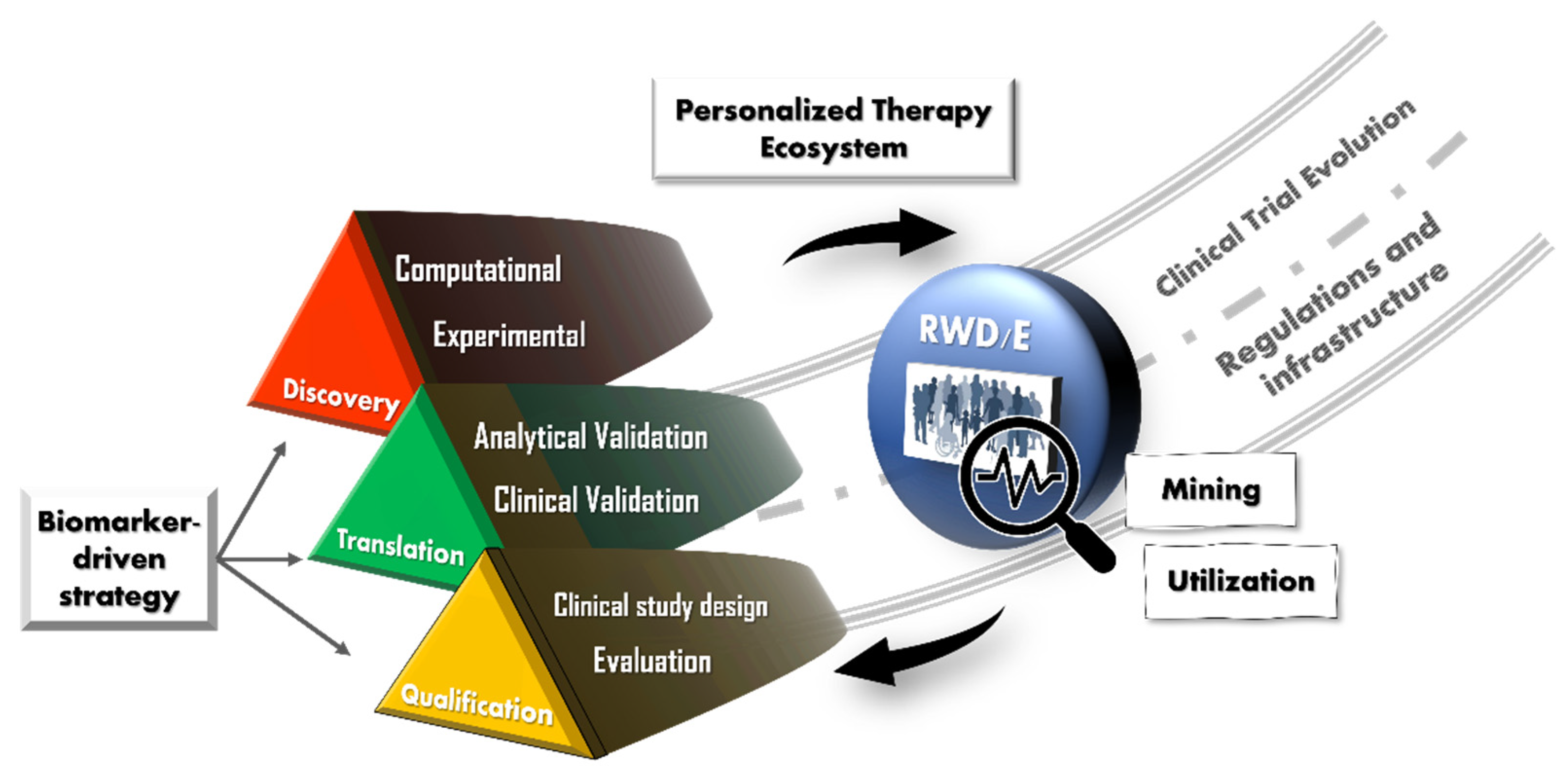
3. Definitions and Scope
4. Discovery
4.1. Big Data and Computational Approaches
4.2. Experimental Approaches
5. Translation
- (1)
- (2)
- Clinical validation/qualification to demonstrate the relationship of a biomarker with the clinical outcome it is posited to be associated with.
6. Qualification
6.1. Mendelian Randomization
6.2. Single Biomarker-Driven Clinical Trials
6.3. Master Protocols and Adaptive Trial Designs
6.4. Evaluating Biomarkers in Personalized Therapies
7. Proposals
7.1. RWD Usage
7.2. Regulations and Infrastructure
7.3. Evolution of the “Clinical Trial”
8. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Food and Drug Administration. FDA Grants Accelerated Approval to Pembrolizumab for First Tissue/Site Agnostic Indication. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pembrolizumab-first-tissuesite-agnostic-indication (accessed on 4 April 2021).
- Frank, R.; Hargreaves, R. Clinical biomarkers in drug discovery and development. Nat. Rev. Drug Discov. 2003, 2, 566–580. [Google Scholar] [CrossRef] [PubMed]
- English, B.; Thomas, K.; Johnstone, J.; Bazih, A.; Gertsik, L.; Ereshefsky, L. Use of translational pharmacodynamic biomarkers in early-phase clinical studies for schizophrenia. Biomark. Med. 2014, 8, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J. The role of biomarkers in Alzheimer’s disease drug development. Adv. Exp. Med. Biol. 2019, 1118, 29–61. [Google Scholar] [PubMed]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef] [Green Version]
- Shema, E.; Bernstein, B.E.; Buenrostro, J.D. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat. Genet. 2018, 51, 19–25. [Google Scholar] [CrossRef]
- Wang, J.; Yang, F. Emerging Single-Cell Technologies for Functional Proteomics in Oncology. Expert Rev. Proteom. 2016, 13, 805–815. [Google Scholar] [CrossRef]
- Song, Y.; Xu, X.; Wang, W.; Tian, T.; Zhu, Z.; Yang, C. Single cell transcriptomics: Moving towards multi-omics. Analyst 2019, 144, 3172–3189. [Google Scholar] [CrossRef]
- Dash, S.; Shakyawar, S.K.; Sharma, M.; Kaushik, S. Big data in healthcare: Management, analysis and future prospects. J. Big Data 2019, 6, 54. [Google Scholar] [CrossRef] [Green Version]
- Camp, J.G.; Platt, R.; Treutlein, B. Mapping human cell phenotypes to genotypes with single-cell genomics. Science 2019, 365, 1401–1405. [Google Scholar] [CrossRef]
- Imai, Y.; Ito, K.; Ueno, T.; Koibuchi, A.; Tanigawa, T.; Serelli-Lee, V.; Matsushima, N. Current status and future prospects in biomarker-driven drug development. In Proceedings of the 42nd Annual Scientific Meeting of the Japanese Society of Clinical Pharmacology and Therapeutics, Sendai, Japan, 9–11 December 2021. [Google Scholar]
- BEST (Biomarkers, EndpointS, and other Tools) Resource. BEST (Biomarkers, EndpointS, and other Tools) Resource 2016. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27010052 (accessed on 15 September 2021).
- Nelson, M.R.; Tipney, H.; Painter, J.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef]
- Morgan, P.; Brown, D.G.; Lennard, S.; Anderton, M.J.; Barrett, J.C.; Eriksson, U.; Fidock, M.; Hamrén, B.; Johnson, A.; March, R.E.; et al. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat. Rev. Drug Discov. 2018, 17, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.S.; Fernando, K.; Allerton, C.; Jansen, K.U.; Vincent, M.S.; Dolsten, M. Reviving an R&D pipeline: A step change in the Phase II success rate. Drug Discov. Today 2020, 26, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. The DNA of a nation. Nature 2015, 524, 503–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogishima, S.; Nagaie, S.; Mizuno, S.; Ishiwata, R.; Iida, K.; Shimokawa, K.; Takai-Igarashi, T.; Nakamura, N.; Nagase, S.; Nakamura, T.; et al. dbTMM: An integrated database of large-scale cohort, genome and clinical data for the Tohoku Medical Megabank Project. Hum. Genome Var. 2021, 8, 1–8. [Google Scholar] [CrossRef]
- Cano-Gamez, E.; Trynka, G. From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases. Front. Genet. 2020, 11, 424. [Google Scholar] [CrossRef]
- Zhou, Y.; Fang, J.; Bekris, L.M.; Kim, Y.H.; Pieper, A.A.; Leverenz, J.B.; Cummings, J.; Cheng, F. AlzGPS: A genome-wide positioning systems platform to catalyze multi-omics for Alzheimer’s drug discovery. Alzheimer’s Res. Ther. 2021, 13, 1–13. [Google Scholar] [CrossRef]
- Leung, E.L.; Cao, Z.-W.; Jiang, Z.-H.; Zhou, H.; Liu, L. Network-based drug discovery by integrating systems biology and computational technologies. Brief. Bioinform. 2012, 14, 491–505. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Kui, L.; Tang, M.; Li, D.; Wei, K.; Chen, W.; Miao, J.; Dong, Y. High-Throughput Transcriptome Profiling in Drug and Biomarker Discovery. Front. Genet. 2020, 11, 19. [Google Scholar] [CrossRef]
- Haymond, A.; Davis, J.B.; Espina, V. Proteomics for cancer drug design. Expert Rev. Proteom. 2019, 16, 647–664. [Google Scholar] [CrossRef]
- Wagatsuma, T.; Nagai-Okatani, C.; Matsuda, A.; Masugi, Y.; Imaoka, M.; Yamazaki, K.; Sakamoto, M.; Kuno, A. Discovery of Pancreatic Ductal Adenocarcinoma-Related Aberrant Glycosylations: A Multilateral Approach of Lectin Microarray-Based Tissue Glycomic Profiling With Public Transcriptomic Datasets. Front. Oncol. 2020, 10, 338. [Google Scholar] [CrossRef] [Green Version]
- Balbas-Martinez, V.; Ruiz-Cerdá, L.; Irurzun-Arana, I.; González-García, I.; Vermeulen, A.; Gómez-Mantilla, J.D.; Trocóniz, I.F. A systems pharmacology model for inflammatory bowel disease. PLoS ONE 2018, 13, e0192949. [Google Scholar] [CrossRef]
- Peskov, K.; Azarov, I.; Chu, L.; Voronova, V.; Kosinsky, Y.; Helmlinger, G. Quantitative Mechanistic Modeling in Support of Pharmacological Therapeutics Development in Immuno-Oncology. Front. Immunol. 2019, 10, 924. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Kim, D.-K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Program (TCGA). Available online: https://www.cancer.gov/tcga (accessed on 24 April 2020).
- Li, X.; Pasche, B.; Zhang, W.; Chen, K. Association of MUC16 Mutation With Tumor Mutation Load and Outcomes in Patients With Gastric Cancer. JAMA Oncol. 2018, 4, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Chen, S.; Gu, L.; Zhang, X. Exploration of Potential Roles of m6A Regulators in Colorectal Cancer Prognosis. Front. Oncol. 2020, 10, 768. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Yu, L.; Cao, Z.; Hu, H.; Luo, S.; Zhang, S. Identification of a Hypoxia-Associated Signature for Lung Adenocarcinoma. Front. Genet. 2020, 11, 647. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, P.G.; Katz, J.P.; Pipas, J.M. Viral sequences in human cancer. Virology 2017, 513, 208–216. [Google Scholar] [CrossRef]
- Chen, L.; Lu, D.; Sun, K.; Xu, Y.; Hu, P.; Li, X.; Xu, F. Identification of biomarkers associated with diagnosis and prognosis of colorectal cancer patients based on integrated bioinformatics analysis. Gene 2019, 692, 119–125. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Nava Rodrigues, D.; Gurel, B.; Clarke, M.; et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Tang, J.; Kong, D.; Cui, Q.; Wang, K.; Zhang, D.; Gong, Y.; Wu, G. Prognostic Genes of Breast Cancer Identified by Gene Co-expression Network Analysis. Front. Oncol. 2018, 8, 374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, X.; Chen, X.; Zhang, Q.; Hong, J. Novel Immune-Related Gene Signature for Risk Stratification and Prognosis of Survival in Lower-Grade Glioma. Front. Genet. 2020, 11, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bando, H. The current status and problems confronted in delivering precision medicine in Japan and Europe. Curr. Probl. Cancer 2017, 41, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Okuma, H.S.; Yonemori, K.; Narita, S.N.; Sukigara, T.; Hirakawa, A.; Shimizu, T.; Shibata, T.; Kawai, A.; Yamamoto, N.; Nakamura, K.; et al. MASTER KEY Project: Powering Clinical Development for Rare Cancers Through a Platform Trial. Clin. Pharmacol. Ther. 2020, 108, 596–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, H.; Matsumoto, S.; Liu, J.; Tanaka, K.; Mori, S.; Hayashi, K.; Kumagai, S.; Shibata, Y.; Hayashida, T.; Watanabe, K.; et al. The CLIP1–LTK fusion is an oncogenic driver in non-small-cell lung cancer. Nature 2021, 600, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Yamaguchi, K.; Urakami, K.; Shimoda, Y.; Ohnami, S.; Ohshima, K.; Tanabe, T.; Naruoka, A.; Kamada, F.; Serizawa, M.; et al. Japanese version of The Cancer Genome Atlas, JCGA, established using fresh frozen tumors obtained from 5143 cancer patients. Cancer Sci. 2019, 111, 687–699. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; De Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Powley, I.R.; Patel, M.; Miles, G.; Pringle, H.; Howells, L.; Thomas, A.; Kettleborough, C.; Bryans, J.; Hammonds, T.; Macfarlane, M.; et al. Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. Br. J. Cancer 2020, 122, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-C.; Jang, S.-Y.; Lee, D.; Lee, J.; Kang, U.; Chang, H.; Kim, H.J.; Han, S.-H.; Seo, J.; Choi, M.; et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat. Commun. 2021, 12, 280. [Google Scholar] [CrossRef]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef] [PubMed]
- National Center Biobank Network. Available online: https://ncbiobank.org/en/home.php (accessed on 31 August 2021).
- Biobank UK. Available online: https://www.ukbiobank.ac.uk/ (accessed on 31 August 2021).
- Biomarker Assay Collaborative Evidentiary Considerations Writing Group. Points to Consider Document: Scientific and Regulatory Considerations for the Analytical Validation of Assays Used in the Qualification of Biomarkers in Biological Matrices; Critical Path Institute: Tuccon, AZ, USA, 2019. [Google Scholar]
- Piccoli, S.; Mehta, D.; Vitaliti, A.; Allinson, J.; Amur, S.; Eck, S.; Green, C.; Hedrick, M.; Hopper, S.; Ji, A.; et al. 2019 White Paper on Recent Issues in Bioanalysis: FDA Immunogenicity Guidance, Gene Therapy, Critical Reagents, Biomarkers and Flow Cytometry Validation (Part 3—Recommendations on 2019 FDA Immunogenicity Guidance, Gene Therapy Bioanalytical Challenges, Strategies for Critical Reagent Management, Biomarker Assay Validation, Flow Cytometry Validation & CLSI H62). Bioanalysis 2019, 11, 2207–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Devanarayan, V.; Barrett, Y.C.; Weiner, R.; Allinson, J.; Fountain, S.; Keller, S.; Weinryb, I.; Green, M.; Duan, L.; et al. Fit-for-Purpose Method Development and Validation for Successful Biomarker Measurement. Pharm. Res. 2006, 23, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research. Biomarker Qualification: Evidentiary Framework. 2018. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/biomarker-qualification-evidentiary-framework (accessed on 15 September 2021).
- Center for Devices and Radiological Health. Principles for Codevelopment of an In Vitro Companion Diagnostic Device with a Therapeutic Product. 2016. Available online: https://www.fda.gov/files/medical%20devices/published/Principles-for-Codevelopment-of-an-In-Vitro-Companion-Diagnostic-Device-with-a-Therapeutic-Product---Draft-Guidance-for-Industry-and-Food-and-Drug-Administration-Staff.pdf (accessed on 15 September 2021).
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Center for Devices and Radiological Health. Use of Real-World Evidence to Support. Regulatory Decision-Making for Medical Devices. 2017. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-real-world-evidence-support-regulatory-decision-making-medical-devices (accessed on 15 September 2021).
- Center for Devices and Radiological Health. Use of Public Human Genetic Variant Databases to Support. Clinical Validity for Genetic and Genomic-Based In Vitro Diagnostics. 2018. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-public-human-genetic-variant-databases-support-clinical-validity-genetic-and-genomic-based-vitro (accessed on 15 September 2021).
- Medical Device Innovation Consortium. Real-World Clinical Evidence Generation: Advancing Regulatory Science and Patient Access for In Vitro Diagnostics (IVDs). 2020. Available online: https://mdic.org/resource/ivd-rwe-framework/ (accessed on 15 September 2021).
- Food & Drug Administration. FDA: MSK-IMPACT Decision Summary. 2017. Available online: https://www.accessdata.fda.gov/cdrh_docs/reviews/DEN170058.pdf (accessed on 15 September 2021).
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, PO.17.00011. [Google Scholar] [CrossRef]
- Lawlor, D.A.; Harbord, R.M.; Sterne, J.A.C.; Timpson, N.; Smith, G.D. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 2008, 27, 1133–1163. [Google Scholar] [CrossRef]
- Yang, Z.; Schooling, C.M.; Kwok, M.K. Mendelian randomization study of interleukin (IL)-1 family and lung cancer. Sci. Rep. 2021, 11, 17606. [Google Scholar] [CrossRef]
- Larsson, S.C.; Traylor, M.; Malik, R.; Dichgans, M.; Burgess, S.; Markus, H.S. Modifiable pathways in Alzheimer’s disease: Mendelian randomisation analysis. BMJ 2017, 359, j5375. [Google Scholar] [CrossRef] [Green Version]
- Howell, A.E.; Zheng, J.; Haycock, P.C.; McAleenan, A.; Relton, C.; Martin, R.M.; Kurian, K.M. Use of Mendelian Randomization for Identifying Risk Factors for Brain Tumors. Front. Genet. 2018, 9, 525. [Google Scholar] [CrossRef]
- E Mokry, L.; Ahmad, O.; Forgetta, V.; Thanassoulis, G.; Richards, J.B. Mendelian randomisation applied to drug development in cardiovascular disease: A review. J. Med. Genet. 2014, 52, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Eiermann, W.; International Herceptin Study Group. International Herceptin Study, Trastuzumab combined with chemotherapy for the treatment of HER2-positive metastatic breast cancer: Pivotal trial data. Ann. Oncol. 2001, 12 (Suppl. 1), S57–S62. [Google Scholar] [CrossRef] [PubMed]
- Freidlin, B.; Korn, E.L. Biomarker enrichment strategies: Matching trial design to biomarker credentials. Nat. Rev. Clin. Oncol. 2013, 11, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.E.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in Previously Treated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Bogin, V. Master protocols: New directions in drug discovery. Contemp. Clin. Trials Commun. 2020, 18, 100568. [Google Scholar] [CrossRef]
- Park, J.J.H.; Siden, E.; Zoratti, M.J.; Dron, L.; Harari, O.; Singer, J.; Lester, R.T.; Thorlund, K.; Mills, E.J. Systematic review of basket trials, umbrella trials, and platform trials: A landscape analysis of master protocols. Trials 2019, 20, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Redman, M.W.; Allegra, C.J. The Master Protocol Concept. Semin. Oncol. 2015, 42, 724–730. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, J.; LaVange, L.M. Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. New Engl. J. Med. 2017, 377, 62–70. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Chronic Pain Master Protocol (CPMP): A Study of LY3016859 in Participants With Osteoarthritis. Available online: https://clinicaltrials.gov/ct2/show/NCT04456686 (accessed on 15 September 2021).
- Derhaschnig, U.; Gilbert, J.; Jäger, U.; Böhmig, G.; Stingl, G.; Jilma, B. Combined integrated protocol/basket trial design for a first-in-human trial. Orphanet J. Rare Dis. 2016, 11, 134. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, T.; Dixon, R.; Page, C.; Carroll, M.; Griffiths, G.; Ho, L.-P.; De Soyza, A.; Felton, T.; Lewis, K.E.; Phekoo, K.; et al. ACCORD: A Multicentre, Seamless, Phase 2 Adaptive Randomisation Platform Study to Assess the Efficacy and Safety of Multiple Candidate Agents for the Treatment of COVID-19 in Hospitalised Patients: A structured summary of a study protocol for a randomised controlled trial. Trials 2020, 21, 1–3. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, S.; Kim, E.S.; Herbst, R.S.; Lee, J.J. Bayesian adaptive design for targeted therapy development in lung cancer—A step toward personalized medicine. Clin. Trials 2008, 5, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Ji, Y.; Catenacci, D.V.T. A subgroup cluster-based Bayesian adaptive design for precision medicine. Biometrics 2016, 73, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.P.; Lindsell, C.J.; Pang, P.S.; Storrow, A.B.; Peacock, W.F.; Levy, P.; Rahbar, M.H.; Del Junco, D.; Gheorghiade, M.; Berry, D.A. Bayesian adaptive trial design in acute heart failure syndromes: Moving beyond the mega trial. Am. Heart J. 2012, 164, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Chen, N.; Wei, C.; Liu, S.; Papadimitrakopoulou, V.A.; Herbst, R.S.; Lee, J.J. Bayesian Two-Stage Biomarker-Based Adaptive Design for Targeted Therapy Development. Stat. Biosci. 2014, 8, 99–128. [Google Scholar] [CrossRef]
- Keeling, P.; Clark, J.; Finucane, S. Challenges in the clinical implementation of precision medicine companion diagnostics. Expert Rev. Mol. Diagn. 2020, 20, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of precision cancer medicine: Evolution of the treatment paradigm. Cancer Treat. Rev. 2020, 86, 102019. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Patel, H.; Nikanjam, M.; Fanta, P.T.; Hahn, M.E.; De, P.; Williams, C.; Guido, J.; et al. Molecular profiling of advanced malignancies guides first-line N-of-1 treatments in the I-PREDICT treatment-naïve study. Genome Med. 2021, 13, 1–14. [Google Scholar] [CrossRef]
- National Cancer Center Japan; Sysmex Corporation; RIKEN GENESIS Co., Ltd. Advanced Medical Care Approval for Cancer Gene Panel Testing at the Time of Initial Treatment—Prospective Study to Assess. the Feasibility and Clinical Utility of Comprehensive Genomic Profiling at the Time of Initial Treatment of Patients with Solid Tumors. 2020. Available online: https://www.ncc.go.jp/en/information/2020/0401/index.html (accessed on 15 September 2021).
- Kasztura, M.; Richard, A.; Bempong, N.-E.; Loncar, D.; Flahault, A. Cost-effectiveness of precision medicine: A scoping review. Int. J. Public Health 2019, 64, 1261–1271. [Google Scholar] [CrossRef] [Green Version]
- Safonov, A.; Wang, S.; Gross, C.P.; Agarwal, D.; Bianchini, G.; Pusztai, L.; Hatzis, C. Assessing cost-utility of predictive biomarkers in oncology: A streamlined approach. Breast Cancer Res. Treat. 2016, 155, 223–234. [Google Scholar] [CrossRef]
- Djalalov, S.; Beca, J.; Hoch, J.S.; Krahn, M.; Tsao, M.-S.; Cutz, J.-C.; Leighl, N.B. Cost Effectiveness of EML4-ALK Fusion Testing and First-Line Crizotinib Treatment for Patients With Advanced ALK-Positive Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2014, 32, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.J.; Canestaro, W.; Ravelo, A.; Wong, W. The cost-effectiveness of alectinib in anaplastic lymphoma kinase-positive (ALK+) advanced NSCLC previously treated with crizotinib. J. Med. Econ. 2017, 20, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Hu, H.; Shi, Y.; She, L.; Yao, L.; Zhu, Y.; Zeng, S.; Shen, L.; Huang, J. Cost-Effectiveness of Pembrolizumab plus Axitinib Versus Sunitinib as First-Line Therapy in Advanced Renal Cell Carcinoma in the U.S. Oncology 2020, 26, e290–e297. [Google Scholar] [CrossRef] [PubMed]
- Jutkowitz, E.; Dubreuil, M.; Lu, N.; Kuntz, K.M.; Choi, H.K. The cost-effectiveness of HLA-B*5801 screening to guide initial urate-lowering therapy for gout in the United States. Semin. Arthritis Rheum. 2016, 46, 594–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, K.; Makimura, T.; Ishiguro, A.; Nonaka, T.; Yamaguchi, M.; Uyama, Y. Evolving Acceptance and Use of RWE for Regulatory Decision Making on the Benefit/Risk Assessment of a Drug in Japan. Clin. Pharmacol. Ther. 2021, 111, 35–43. [Google Scholar] [CrossRef]
- Mukai, Y.; Ueno, H. Establishment and implementation of Cancer Genomic Medicine in Japan. Cancer Sci. 2020, 112, 970–977. [Google Scholar] [CrossRef]
- Furusawa, Y.; Yamaguchi, I.; Yagishita, N.; Tanzawa, K.; Matsuda, F.; Yamano, Y. RADDAR-J Research and Development Group National platform for Rare Diseases Data Registry of Japan. Learn. Health Syst. 2019, 3, e10080. [Google Scholar] [CrossRef]
- Cirillo, D.; Valencia, A. Big data analytics for personalized medicine. Curr. Opin. Biotechnol. 2019, 58, 161–167. [Google Scholar] [CrossRef]
- Nagai, S.; Urata, M.; Sato, H.; Mikami, M.; Kuga, W.; Yanagihara, R.; Miyamoto, D.; Suzuki, Y.; Shikano, M. Evolving Japanese regulations on companion diagnostics. Nat. Biotechnol. 2016, 34, 141–144. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kanayama, N.; Nakayama, Y.; Matsushima, N. Current Status, Issues and Future Prospects of Personalized Medicine for Each Disease. J. Pers. Med. 2022, 12, 444. [Google Scholar] [CrossRef]
- Schuhmacher, A.; Wilisch, L.; Kuss, M.; Kandelbauer, A.; Hinder, M.; Gassmann, O. R&D efficiency of leading pharmaceutical companies—A 20-year analysis. Drug Discov. Today 2021, 26, 1784–1789. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.-P.; Zhang, X.; Aronov, A.M. Drug discovery effectiveness from the standpoint of therapeutic mechanisms and indications. Nat. Rev. Drug Discov. 2017, 17, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Dancey, J.E.; Dobbin, K.K.; Groshen, S.; Jessup, J.M.; Hruszkewycz, A.H.; Koehler, M.; Parchment, R.; Ratain, M.J.; Shankar, L.K.; Stadler, W.M.; et al. Guidelines for the Development and Incorporation of Biomarker Studies in Early Clinical Trials of Novel Agents. Clin. Cancer Res. 2010, 16, 1745–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Approach | Specific Method | Source of Data/Samples | Applications in Drug Development |
|---|---|---|---|
| Computational |
|
|
|
| Experimental |
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serelli-Lee, V.; Ito, K.; Koibuchi, A.; Tanigawa, T.; Ueno, T.; Matsushima, N.; Imai, Y. A State-of-the-Art Roadmap for Biomarker-Driven Drug Development in the Era of Personalized Therapies. J. Pers. Med. 2022, 12, 669. https://doi.org/10.3390/jpm12050669
Serelli-Lee V, Ito K, Koibuchi A, Tanigawa T, Ueno T, Matsushima N, Imai Y. A State-of-the-Art Roadmap for Biomarker-Driven Drug Development in the Era of Personalized Therapies. Journal of Personalized Medicine. 2022; 12(5):669. https://doi.org/10.3390/jpm12050669
Chicago/Turabian StyleSerelli-Lee, Victoria, Kazumi Ito, Akira Koibuchi, Takahiko Tanigawa, Takayo Ueno, Nobuko Matsushima, and Yasuhiko Imai. 2022. "A State-of-the-Art Roadmap for Biomarker-Driven Drug Development in the Era of Personalized Therapies" Journal of Personalized Medicine 12, no. 5: 669. https://doi.org/10.3390/jpm12050669
APA StyleSerelli-Lee, V., Ito, K., Koibuchi, A., Tanigawa, T., Ueno, T., Matsushima, N., & Imai, Y. (2022). A State-of-the-Art Roadmap for Biomarker-Driven Drug Development in the Era of Personalized Therapies. Journal of Personalized Medicine, 12(5), 669. https://doi.org/10.3390/jpm12050669