
Mechanistic Insights in Hemophagocytic Lymphohistiocytosis: Subsequent Acute Hepatic Failure in a Multiple Myeloma Patient Following Therapy with Ixazomib-Lenalidomide-Dexamethasone

 , ,
, ,
Abstract
:1. Background and Nomenclature
- (1)
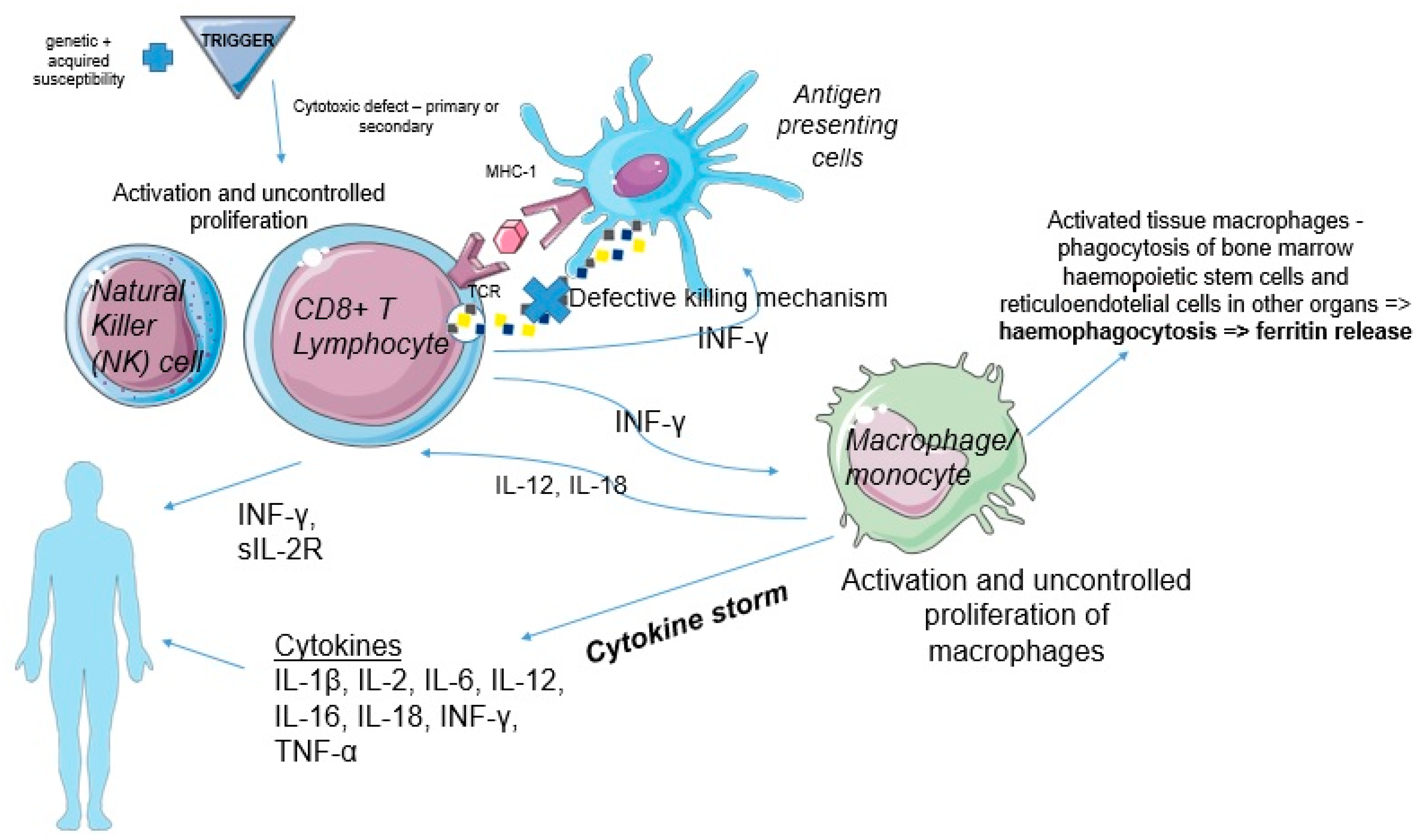
- HLH syndrome refers to patients who meet consensus diagnostic criteria. The immune activation is often associated with lymphocyte cytotoxicity due to genetic defects;
- (2)
- HLH disease: an HLH syndrome in which the distinctive immune dysregulation is the core problem and in which immune suppression could be of benefit. HLH disease may be associated with specific genetic and/or environmental causes.HLH disease can be:
- -
- Familial HLH with clear genetic etiology (F-HLH);
- -
- HLH associated with malignancy (M-HLH);
- -
- HLH associated with rheumatologic conditions (MAS) (R-HLH);
- -
- HLH observed after immune-activating therapies (iatrogenic HLH, also called cytokine release syndrome) (Rx-HLH);
- -
- HLH associated with immune compromise (either primary immune deficiency or treatment-related immune suppression) (IC-HLH);
- -
- HLH not associated with other specific conditions (those with negative or ambiguous genetic findings, with or without infectious triggers) (HLH-disease NOS).
- (3)
- HLH disease mimics: disorders that follow the same pattern as HLH syndrome but are caused by other conditions and which might not benefit from immune suppression [12].
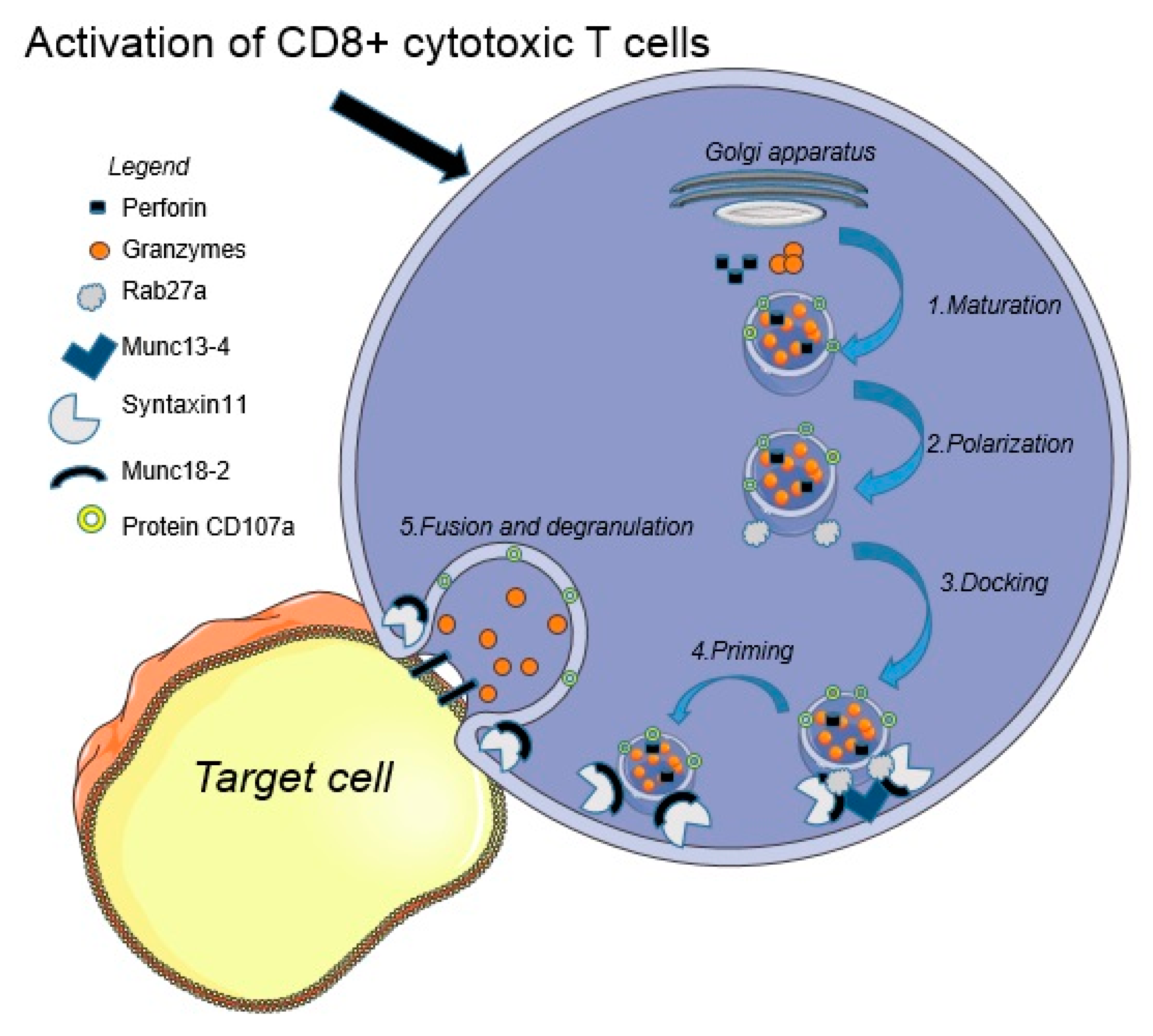
2. Pathogenesis
3. Clinical Manifestations
4. Diagnosis and Differential Diagnosis
5. Current Treatment Strategies and Future Approaches
6. Prognosis
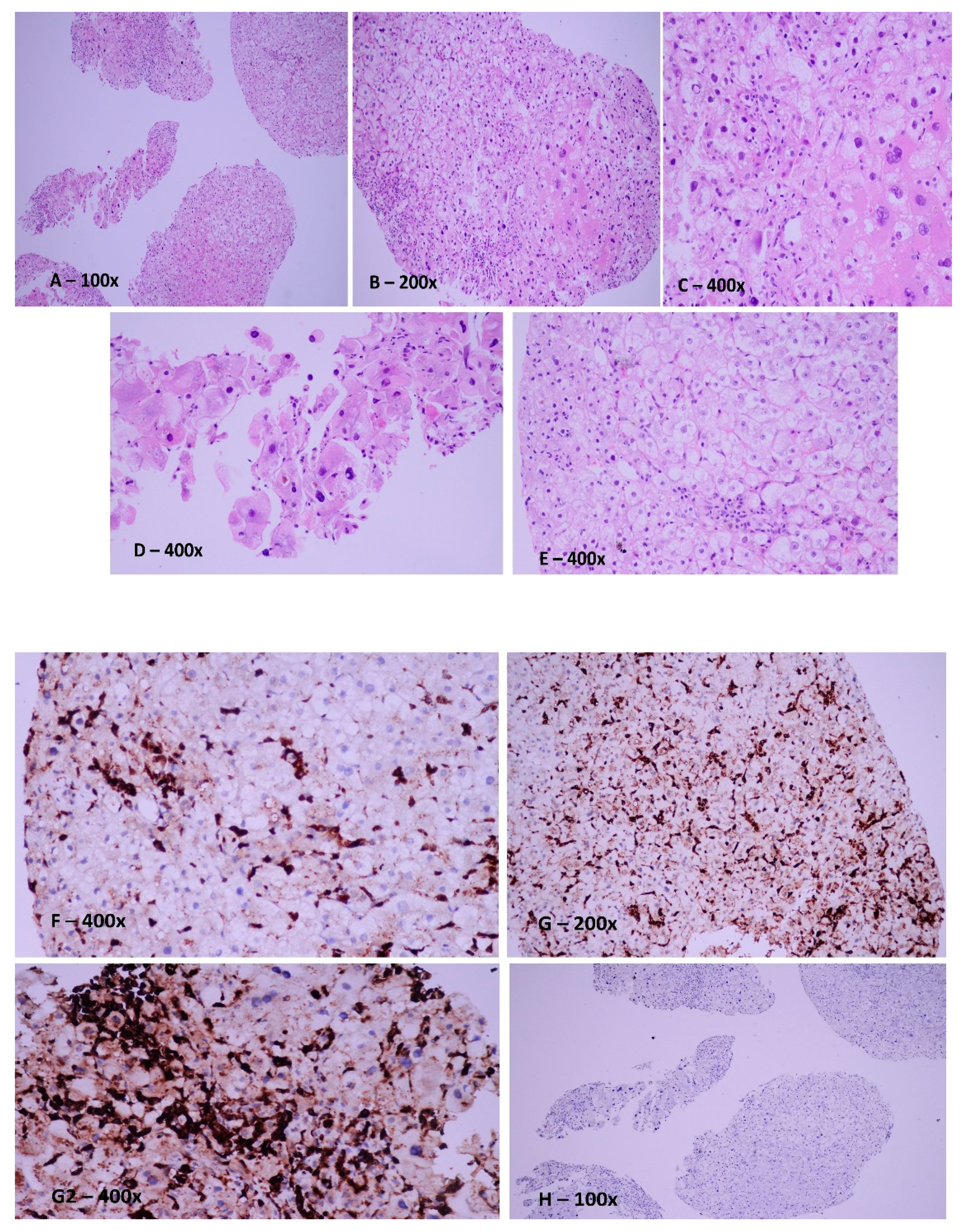
7. Case Report
8. Discussions
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Samkari, H.; Berliner, N. Hemophagocytic Lymphohistiocytosis. Annu. Rev. Pathol. 2018, 13, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Aricò, M.; Janka, G.; Fischer, A.; Henter, J.I.; Blanche, S.; Elinder, G.; Martinetti, M.; Rusca, M.P. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia 1996, 10, 197–203. [Google Scholar] [PubMed]
- Henter, J.-I.; Samuelsson-Horne, A.; Aricò, M.; Egeler, R.M.; Elinder, G.; Filipovich, A.H.; Gadner, H.; Imashuku, S.; Komp, D.; Ladisch, S.; et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood 2002, 100, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brito-Zerón, P.; López-Guillermo, A.; Khamashta, M.A.; Bosch, X. Adult haemophagocytic syndrome. Lancet 2014, 383, 1503–1516. [Google Scholar] [CrossRef]
- Griffin, G.; Shenoi, S.; Hughes, G.C. Hemophagocytic lymphohistiocytosis: An update on pathogenesis, diagnosis, and therapy. Best Pract. Res. Clin. Rheumatol. 2020, 34, 101515. [Google Scholar] [CrossRef]
- La Rosée, P.; Horne, A.; Hines, M.; von Bahr Greenwood, T.; Machowicz, R.; Berliner, N.; Birndt, S.; Gil-Herrera, J.; Girschikofsky, M.; Jordan, M.B.; et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood 2019, 133, 2465–2477. [Google Scholar] [CrossRef] [Green Version]
- Wegehaupt, O.; Wustrau, K.; Lehmberg, K.; Ehl, S. Cell Versus Cytokine—Directed Therapies for Hemophagocytic Lymphohistiocytosis (HLH) in Inborn Errors of Immunity. Front. Immunol. 2020, 11, 808. [Google Scholar] [CrossRef]
- Rouphael, N.G.; Talati, N.J.; Vaughan, C.; Cunningham, K.; Moreira, R.; Gould, C. Infections associated with haemophagocytic syndrome. Lancet Infect. Dis. 2007, 7, 814–822. [Google Scholar] [CrossRef]
- Lehmberg, K.; Nichols, K.E.; Henter, J.-I.; Girschikofsky, M.; Greenwood, T.; Jordan, M.; Kumar, A.; Minkov, M.; La Rosée, P.; Weitzman, S.; et al. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica 2015, 100, 997–1004. [Google Scholar] [CrossRef]
- Carter, S.J.; Tattersall, R.S.; Ramanan, A.V. Macrophage activation syndrome in adults: Recent advances in pathophysiology, diagnosis and treatment. Rheumatology 2019, 58, 5–17. [Google Scholar] [CrossRef]
- Asano, T.; Kogawa, K.; Morimoto, A.; Ishida, Y.; Suzuki, N.; Ohga, S.; Kudo, K.; Ohta, S.; Wakiguchi, H.; Tabuchi, K.; et al. Hemophagocytic lymphohistiocytosis after hematopoietic stem cell transplantation in children: A nationwide survey in Japan. Pediatr. Blood Cancer 2012, 59, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.B.; Allen, C.E.; Greenberg, J.; Henry, M.; Hermiston, M.L.; Kumar, A.; Hines, M.; Eckstein, O.; Ladisch, S.; Nichols, K.E.; et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr. Blood Cancer 2019, 66, e27929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisse, E.; Wouters, C.H.; Matthys, P. Hemophagocytic lymphohistiocytosis (HLH): A heterogeneous spectrum of cytokine-driven immune disorders. Cytokine Growth Factor Rev. 2015, 26, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Widowati, W.; Jasaputra, D.K.; Sumitro, S.B.; Widodo, M.A.; Mozef, T.; Rizal, R.; Kusuma, H.S.W.; Laksmitawati, D.R.; Murti, H.; Bachtiar, I.; et al. Effect of interleukins (IL-2, IL-15, IL-18) on receptors activation and cytotoxic activity of natural killer cells in breast cancer cell. Afr. Health Sci. 2020, 20, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Aktas, E.; Kucuksezer, U.C.; Bilgic, S.; Erten, G.; Deniz, G. Relationship between CD107a expression and cytotoxic activity. Cell Immunol. 2009, 254, 149–154. [Google Scholar] [CrossRef]
- Filipovich, A.H.; Chandrakasan, S. Pathogenesis of Hemophagocytic Lymphohistiocytosis. Hematol. Oncol. Clin. N. Am. 2015, 29, 895–902. [Google Scholar] [CrossRef]
- Bordbar, M.R.; Modarresi, F.; Farazi Fard, M.A.; Dastsooz, H.; Shakib Azad, N.; Faghihi, M.A. A case report of novel mutation in PRF1 gene, which causes familial autosomal recessive hemophagocytic lymphohistiocytosis. BMC Med. Genet. 2017, 18, 49. [Google Scholar] [CrossRef] [Green Version]
- de Saint Basile, G.; Ménasché, G.; Fischer, A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat. Rev. Immunol. 2010, 10, 568–579. [Google Scholar] [CrossRef]
- Giri, P.P.; Biswas, N.; Chakravarty, S. Familial Hemophagocytic Lymphohistiocytosis due to Mutation of UNC13D Gene. Indian J. Hematol. Blood Transfus. 2016, 32, 344–346. [Google Scholar] [CrossRef]
- Marsh, R.A.; Satake, N.; Biroschak, J.; Jacobs, T.; Johnson, J.; Jordan, M.B.; Bleesing, J.J.; Filipovich, A.H.; Zhang, K. STX11 mutations and clinical phenotypes of familial hemophagocytic lymphohistiocytosis in North America. Pediatr. Blood Cancer 2010, 55, 134–140. [Google Scholar] [CrossRef]
- Pachlopnik Schmid, J.; Schmid, J.P.; Côte, M.; Ménager, M.M.; Burgess, A.; Nehme, N.; Ménasché, G.; Fischer, A.; de Saint Basile, G. Inherited defects in lymphocyte cytotoxic activity. Immunol. Rev. 2010, 235, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Esmaeilzadeh, H.; Bemanian, M.H.; Nabavi, M.; Arshi, S.; Fallahpour, M.; Fuchs, I.; zur Stadt, U.; Warnatz, K.; Ammann, S.; Ehl, S.; et al. Novel Patient with Late-Onset Familial Hemophagocytic Lymphohistiocytosis with STXBP2 Mutations Presenting with Autoimmune Hepatitis, Neurological Manifestations and Infections Associated with Hypogammaglobulinemia. J. Clin. Immunol. 2015, 35, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Canna, S.W.; Marsh, R.A. Pediatric hemophagocytic lymphohistiocytosis. Blood 2020, 135, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Bracaglia, C.; Prencipe, G.; Bemrich-Stolz, C.J.; Beukelman, T.; Dimmitt, R.A.; Chatham, W.W.; Zhang, K.; Li, H.; Walter, M.R.; et al. A Heterozygous RAB27A Mutation Associated with Delayed Cytolytic Granule Polarization and Hemophagocytic Lymphohistiocytosis. J. Immunol. 2016, 196, 2492–2503. [Google Scholar] [CrossRef] [Green Version]
- Barsalou, J.; Blincoe, A.; Fernandez, I.; Dal-Soglio, D.; Marchitto, L.; Selleri, S.; Haddad, E.; Benyoucef, A.; Touzot, F. Rapamycin as an Adjunctive Therapy for NLRC4 Associated Macrophage Activation Syndrome. Front. Immunol. 2018, 9, 2162. [Google Scholar] [CrossRef]
- Latour, S.; Fischer, A. Signaling pathways involved in the T-cell-mediated immunity against Epstein-Barr virus: Lessons from genetic diseases. Immunol. Rev. 2019, 291, 174–189. [Google Scholar] [CrossRef]
- Chinn, I.K.; Eckstein, O.S.; Peckham-Gregory, E.C.; Goldberg, B.R.; Forbes, L.R.; Nicholas, S.K.; Mace, E.M.; Vogel, T.P.; Abhyankar, H.A.; Diaz, M.I.; et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood 2018, 132, 89–100. [Google Scholar] [CrossRef]
- Jordan, M.B.; Allen, C.E.; Weitzman, S.; Filipovich, A.H.; McClain, K.L. How I treat hemophagocytic lymphohistiocytosis. Blood 2011, 118, 4041–4052. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, F.; Hibi, S.; Imashuku, S. Hypercytokinemia in hemophagocytic syndrome. Am. J. Pediatr. Hematol. Oncol. 1993, 15, 92–98. [Google Scholar] [CrossRef]
- Takada, H.; Nomura, A.; Ohga, S.; Hara, T. Interleukin-18 in hemophagocytic lymphohistiocytosis. Leuk. Lymphoma 2001, 42, 21–28. [Google Scholar] [CrossRef]
- Crayne, C.B.; Albeituni, S.; Nichols, K.E.; Cron, R.Q. The Immunology of Macrophage Activation Syndrome. Front. Immunol. 2019, 10, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billiau, A.D.; Roskams, T.; Van Damme-Lombaerts, R.; Matthys, P.; Wouters, C. Macrophage activation syndrome: Characteristic findings on liver biopsy illustrating the key role of activated, IFN-gamma-producing lymphocytes and IL-6- and TNF-alpha-producing macrophages. Blood 2005, 105, 1648–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gars, E.; Purington, N.; Scott, G.; Chisholm, K.; Gratzinger, D.; Martin, B.A.; Ohgami, R.S. Bone marrow histomorphological criteria can accurately diagnose hemophagocytic lymphohistiocytosis. Haematologica 2018, 103, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Girard-Guyonvarc’h, C.; Palomo, J.; Martin, P.; Rodriguez, E.; Troccaz, S.; Palmer, G.; Gabay, C. Unopposed IL-18 signaling leads to severe TLR9-induced macrophage activation syndrome in mice. Blood 2018, 131, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- Janka, G.E.; Lehmberg, K. Hemophagocytic lymphohistiocytosis: Pathogenesis and treatment. Hematology Am. Soc. Hematol. Educ. Program 2013, 2013, 605–611. [Google Scholar] [CrossRef] [Green Version]
- Palazzi, D.L.; McClain, K.L.; Kaplan, S.L. Hemophagocytic syndrome in children: An important diagnostic consideration in fever of unknown origin. Clin. Infect. Dis. 2003, 36, 306–312. [Google Scholar] [CrossRef]
- Nikiforow, S.; Berliner, N. The unique aspects of presentation and diagnosis of hemophagocytic lymphohistiocytosis in adults. Hematology Am. Soc. Hematol. Educ. Program 2015, 2015, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Henter, J.-I.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2007, 48, 124–131. [Google Scholar] [CrossRef]
- Locatelli, F.; Jordan, M.B.; Allen, C.; Cesaro, S.; Rizzari, C.; Rao, A.; Degar, B.; Garrington, T.P.; Sevilla, J.; Putti, M.-C.; et al. Emapalumab in Children with Primary Hemophagocytic Lymphohistiocytosis. N. Engl. J. Med. 2020, 382, 1811–1822. [Google Scholar] [CrossRef]
- Schram, A.M.; Berliner, N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 2015, 125, 2908–2914. [Google Scholar] [CrossRef]
- de Kerguenec, C.; Hillaire, S.; Molinié, V.; Gardin, C.; Degott, C.; Erlinger, S.; Valla, D. Hepatic manifestations of hemophagocytic syndrome: A study of 30 cases. Am. J. Gastroenterol. 2001, 96, 852–857. [Google Scholar] [CrossRef]
- Fukaya, S.; Yasuda, S.; Hashimoto, T.; Oku, K.; Kataoka, H.; Horita, T.; Atsumi, T.; Koike, T. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: Analysis of 30 cases. Rheumatology 2008, 47, 1686–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ost, A.; Nilsson-Ardnor, S.; Henter, J.I. Autopsy findings in 27 children with haemophagocytic lymphohistiocytosis. Histopathology 1998, 32, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Bergsten, E.; Horne, A.; Aricó, M.; Astigarraga, I.; Egeler, R.M.; Filipovich, A.H.; Ishii, E.; Janka, G.; Ladisch, S.; Lehmberg, K.; et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: Long-term results of the cooperative HLH-2004 study. Blood 2017, 130, 2728–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schram, A.M.; Campigotto, F.; Mullally, A.; Fogerty, A.; Massarotti, E.; Neuberg, D.; Berliner, N. Marked hyperferritinemia does not predict for HLH in the adult population. Blood 2015, 125, 1548–1552. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.E.; Yu, X.; Kozinetz, C.A.; McClain, K.L. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2008, 50, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, A.; Nakazawa, Y.; Ishii, E. Hemophagocytic lymphohistiocytosis: Pathogenesis, diagnosis, and management. Pediatr. Int. 2016, 58, 817–825. [Google Scholar] [CrossRef]
- Wu, J.-R.; Yuan, L.-X.; Ma, Z.-G.; Chen, X.-X.; Gu, L.; Gao, J. GDF15-mediated upregulation of ferroportin plays a key role in the development of hyperferritinemia in children with hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2013, 60, 940–945. [Google Scholar] [CrossRef]
- Lin, M.; Park, S.; Hayden, A.; Giustini, D.; Trinkaus, M.; Pudek, M.; Mattman, A.; Schneider, M.; Chen, L.Y.C. Clinical utility of soluble interleukin-2 receptor in hemophagocytic syndromes: A systematic scoping review. Ann. Hematol. 2017, 96, 1241–1251. [Google Scholar] [CrossRef]
- Fardet, L.; Galicier, L.; Lambotte, O.; Marzac, C.; Aumont, C.; Chahwan, D.; Coppo, P.; Hejblum, G. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014, 66, 2613–2620. [Google Scholar] [CrossRef]
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med. 2017, 43, 304–377. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. HLH Across Speciality Collaboration, UK COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Dwyre, D.M.; Holland, P.V. Transfusion-associated graft-versus-host disease. Vox Sang. 2008, 95, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, V.; Kolli, S.S.; Strowd, L.C. Review of Graft-Versus-Host Disease. Dermatol Clin. 2019, 37, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-J.; Tang, Y.-M.; Song, H.; Yang, S.-L.; Xu, W.-Q.; Zhao, N.; Shi, S.-W.; Shen, H.-P.; Mao, J.-Q.; Zhang, L.-Y.; et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J. Pediatr. 2012, 160, 984–990.e1. [Google Scholar] [CrossRef]
- Constantinescu, C.; Pasca, S.; Tat, T.; Teodorescu, P.; Vlad, C.; Iluta, S.; Dima, D.; Tomescu, D.; Scarlatescu, E.; Tanase, A.; et al. Continuous renal replacement therapy in cytokine release syndrome following immunotherapy or cellular therapies? J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Allen, C.E.; McClain, K.L. Pathophysiology and epidemiology of hemophagocytic lymphohistiocytosis. Hematol. Am. Soc. Hematol. Educ. Program 2015, 2015, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Lehmberg, K.; Sprekels, B.; Nichols, K.E.; Woessmann, W.; Müller, I.; Suttorp, M.; Bernig, T.; Beutel, K.; Bode, S.F.N.; Kentouche, K.; et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br. J. Haematol. 2015, 170, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Machaczka, M.; Vaktnäs, J.; Klimkowska, M.; Hägglund, H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: A retrospective population-based analysis from a single center. Leuk. Lymphoma 2011, 52, 613–619. [Google Scholar] [CrossRef]
- Strenger, V.; Merth, G.; Lackner, H.; Aberle, S.W.; Kessler, H.H.; Seidel, M.G.; Schwinger, W.; Sperl, D.; Sovinz, P.; Karastaneva, A.; et al. Malignancy and chemotherapy induced haemophagocytic lymphohistiocytosis in children and adolescents—A single centre experience of 20 years. Ann. Hematol. 2018, 97, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Barrett, D.; Teachey, D.T.; Grupp, S.A. Managing Cytokine Release Syndrome Associated With Novel T Cell-Engaging Therapies. Cancer J. 2014, 20, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Tomuleasa, C.; Fuji, S.; Berce, C.; Onaciu, A.; Chira, S.; Petrushev, B.; Micu, W.-T.; Moisoiu, V.; Osan, C.; Constantinescu, C.; et al. Chimeric Antigen Receptor T-Cells for the Treatment of B-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2018, 9, 239. [Google Scholar] [CrossRef] [PubMed]
- Ehl, S.; Astigarraga, I.; von Bahr Greenwood, T.; Hines, M.; Horne, A.; Ishii, E.; Janka, G.; Jordan, M.B.; La Rosée, P.; Lehmberg, K.; et al. Recommendations for the Use of Etoposide-Based Therapy and Bone Marrow Transplantation for the Treatment of HLH: Consensus Statements by the HLH Steering Committee of the Histiocyte Society. J. Allergy Clin. Immunol. Pract. 2018, 6, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Merrill, S.A.; Alsawah, F.; Bockenstedt, P.; Campagnaro, E.; Devata, S.; Gitlin, S.D.; Kaminski, M.; Cusick, A.; Phillips, T.; et al. Ruxolitinib in adult patients with secondary haemophagocytic lymphohistiocytosis: An open-label, single-centre, pilot trial. Lancet Haematol. 2019, 6, e630–e637. [Google Scholar] [CrossRef]
- Marsh, R.A.; Jordan, M.B.; Talano, J.-A.; Nichols, K.E.; Kumar, A.; Naqvi, A.; Vaiselbuh, S.R. Histiocyte Society Salvage Therapy Working Group Salvage therapy for refractory hemophagocytic lymphohistiocytosis: A review of the published experience. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef]
- Jabado, N.; de Graeff-Meeder, E.R.; Cavazzana-Calvo, M.; Haddad, E.; Le Deist, F.; Benkerrou, M.; Dufourcq, R.; Caillat, S.; Blanche, S.; Fischer, A. Treatment of familial hemophagocytic lymphohistiocytosis with bone marrow transplantation from HLA genetically nonidentical donors. Blood 1997, 90, 4743–4748. [Google Scholar] [CrossRef] [Green Version]
- Marsh, R.A.; Vaughn, G.; Kim, M.-O.; Li, D.; Jodele, S.; Joshi, S.; Mehta, P.A.; Davies, S.M.; Jordan, M.B.; Bleesing, J.J.; et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood 2010, 116, 5824–5831. [Google Scholar] [CrossRef] [Green Version]
- Ravelli, A.; Davì, S.; Minoia, F.; Martini, A.; Cron, R.Q. Macrophage Activation Syndrome. Hematol. Oncol. Clin. N. Am. 2015, 29, 927–941. [Google Scholar] [CrossRef]
- Al-Salama, Z.T. Emapalumab: First Global Approval. Drugs 2019, 79, 99–103. [Google Scholar] [CrossRef]
- Booth, C.; Carmo, M.; Gaspar, H.B. Gene therapy for haemophagocytic lymphohistiocytosis. Curr. Gene Ther. 2014, 14, 437–446. [Google Scholar] [CrossRef]
- Janka, G.E. Familial hemophagocytic lymphohistiocytosis. Eur. J. Pediatr. 1983, 140, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Trottestam, H.; Horne, A.; Aricò, M.; Egeler, R.M.; Filipovich, A.H.; Gadner, H.; Imashuku, S.; Ladisch, S.; Webb, D.; Janka, G.; et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: Long-term results of the HLH-94 treatment protocol. Blood 2011, 118, 4577–4584. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Comstock, P.; Campo, M.; Gorovets, D.; Mullally, A.; Bodio, K.; Arnason, J.; Berliner, N. Haemophagocytic lymphohistiocytosis in adults: A multicentre case series over 7 years. Br. J. Haematol. 2016, 172, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Terrovitis, J.V.; Matsouka, C.; Anagnostopoulos, A.; Anastasiou-Nana, M.I.; Dimopoulos, A.M. Hemophagocytic lymphohistiocytosis after chemotherapy for multiple myeloma. Clin. Lymphoma 2004, 5, 194–196. [Google Scholar] [CrossRef]
- Nagafuji, K.; Nonami, A.; Kumano, T.; Kikushige, Y.; Yoshimoto, G.; Takenaka, K.; Shimoda, K.; Ohga, S.; Yasukawa, M.; Horiuchi, H.; et al. Perforin gene mutations in adult-onset hemophagocytic lymphohistiocytosis. Haematologica 2007, 92, 978–981. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Primary HLH (Mendelian Inherited Conditions) [7] | Secondary HLH |
|---|---|
| Defects in the cytolytic function of cytotoxic T cells and/or NK cells | Infections (EBV, HIV, CMV, SARS-CoV-2, bacterial, fungi, parasites) [8] |
| Defects in inflammasome regulation | Malignancies (lymphomas) T-cell or natural killer (NK) cell lymphomas, B-cell lymphomas, leukemias, Hodgkin lymphoma, solid tumors (all require a meticulous search for the underlying disease) [9] |
| MAS or autoimmune disorders: systemic-onset juvenile idiopathic arthritis (sJIA), adult-onset Still’s disease (ASD), vasculitis, systemic lupus erythematosus (LES) [10] | |
| Organ (kidney) or stem cell transplantation [11] | |
| Metabolic, surgery, trauma | |
| Immunosuppression, vaccination, hemodialysis, immune-activating therapy (e.g., CAR-T therapy) | |
| Pregnancy |
| The Diagnosis of HLH Can Be Established If Criterion 1 or 2 are Fulfilled. |
|---|
| 1. A molecular diagnosis consistent with HLH: pathologic mutations of PRF1, UNC13D, Munc18-2, Rab27a, STX11, SH2D1A, or BIRC4 |
| 2. Diagnostic criteria for HLH fulfilled (≥5 of the 8 criteria below) |
| (1) Fever ≥ 38.5 °C |
| (2) Splenomegaly |
| (3) Cytopenias (affecting ≥2 of 3 lineages in the peripheral blood) |
| Hemoglobin < 9 g/dL (hemoglobin < 10 g/dL in infants < 4 weeks) |
| Platelets < 100 × 103/mL |
| Neutrophils < 1 × 103/mL |
| (4) Hypertriglyceridemia and/or hypofibrinogenemia |
| Fasting triglycerides ≥ 3.0 mmol/L (i.e., ≥265 mg/dL) |
| Fibrinogen ≤ 150 mg/dL |
| (5) Hemophagocytosis in bone marrow, spleen, liver, or lymph nodes with no evidence of malignancy. |
| (6) Low or no NK cell activity (according to local laboratory reference) |
| (7) Ferritin ≥ 500 ng/mL |
| (8) sCD25 (i.e., α-chain soluble IL-2 receptor) ≥ 2400 U/mL correlated with current disease activity [28] |
| (9) Elevated CXCL9 [39] (not in the original classification criteria) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Constantinescu, C.; Petrushev, B.; Rus, I.; Stefanescu, H.; Frasinariu, O.; Margarit, S.; Dima, D.; Tomuleasa, C. Mechanistic Insights in Hemophagocytic Lymphohistiocytosis: Subsequent Acute Hepatic Failure in a Multiple Myeloma Patient Following Therapy with Ixazomib-Lenalidomide-Dexamethasone. J. Pers. Med. 2022, 12, 678. https://doi.org/10.3390/jpm12050678
Constantinescu C, Petrushev B, Rus I, Stefanescu H, Frasinariu O, Margarit S, Dima D, Tomuleasa C. Mechanistic Insights in Hemophagocytic Lymphohistiocytosis: Subsequent Acute Hepatic Failure in a Multiple Myeloma Patient Following Therapy with Ixazomib-Lenalidomide-Dexamethasone. Journal of Personalized Medicine. 2022; 12(5):678. https://doi.org/10.3390/jpm12050678
Chicago/Turabian StyleConstantinescu, Catalin, Bobe Petrushev, Ioana Rus, Horia Stefanescu, Otilia Frasinariu, Simona Margarit, Delia Dima, and Ciprian Tomuleasa. 2022. "Mechanistic Insights in Hemophagocytic Lymphohistiocytosis: Subsequent Acute Hepatic Failure in a Multiple Myeloma Patient Following Therapy with Ixazomib-Lenalidomide-Dexamethasone" Journal of Personalized Medicine 12, no. 5: 678. https://doi.org/10.3390/jpm12050678