
Signal-to-Noise Analysis Can Inform the Likelihood That Incidentally Identified Variants in Sarcomeric Genes Are Associated with Pediatric Cardiomyopathy

 , ,
, ,
Abstract
:1. Introduction
2. Methods
3. Results
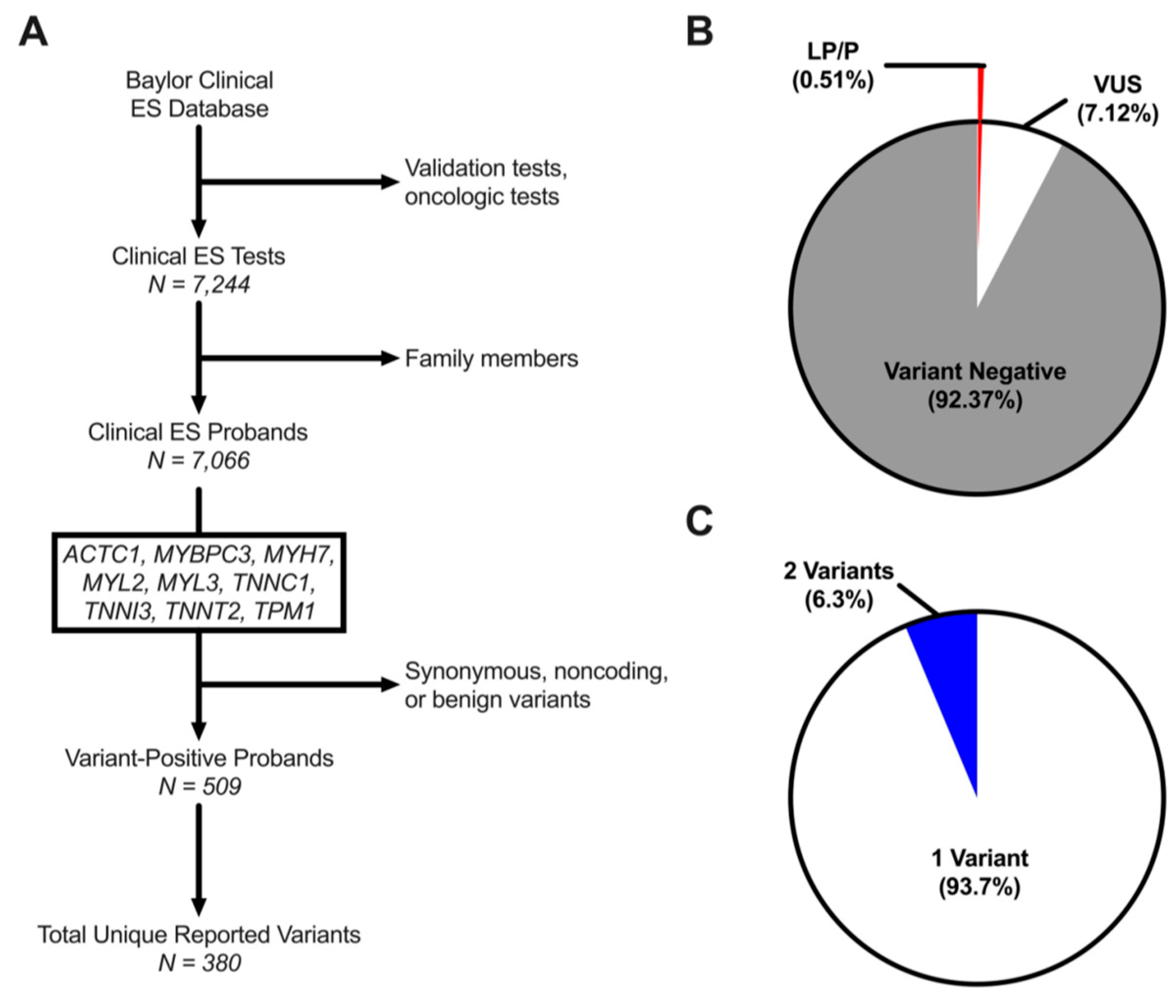
3.1. Rare Variant Prevalence in HCM-Associated Genes among ES Referrals
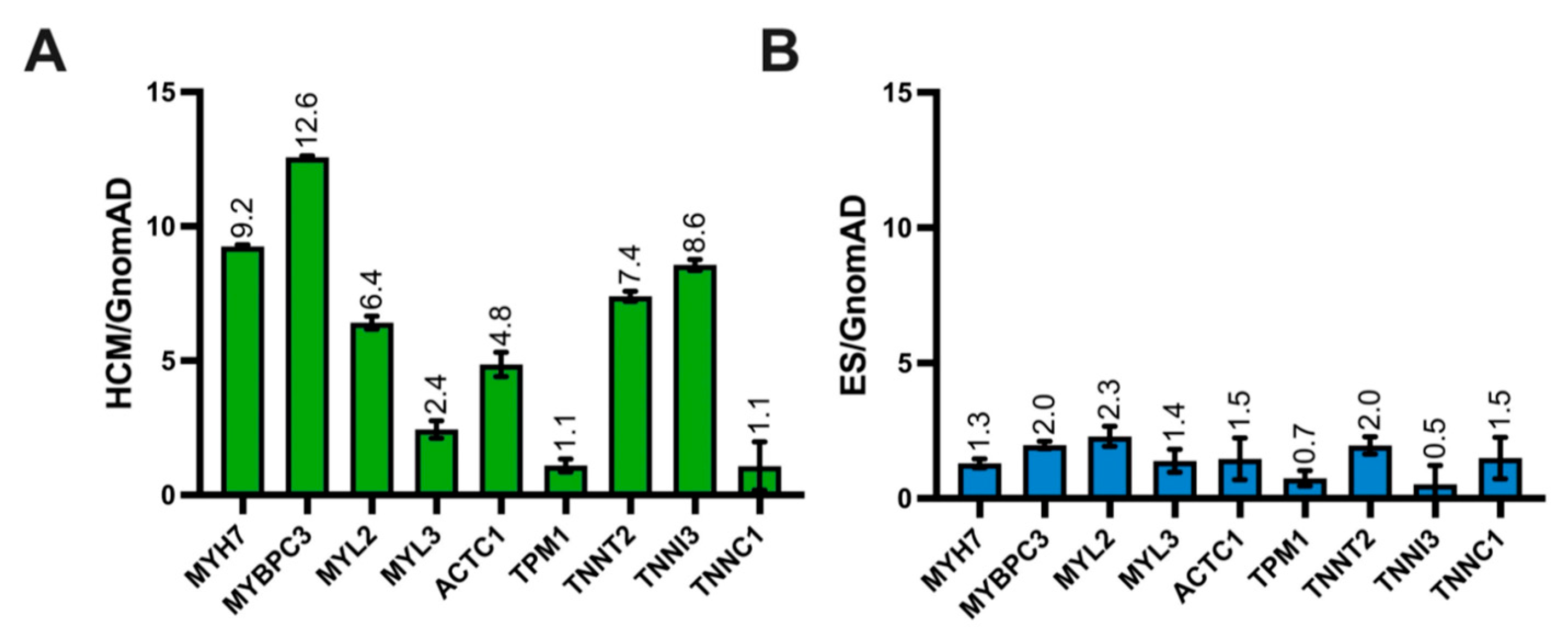
3.2. ES Variant Frequency Compared to Control and HCM-Afflicted Individuals
3.3. Gene-Level Signal-to-Noise in ES and Pathogenic HCM Cohort
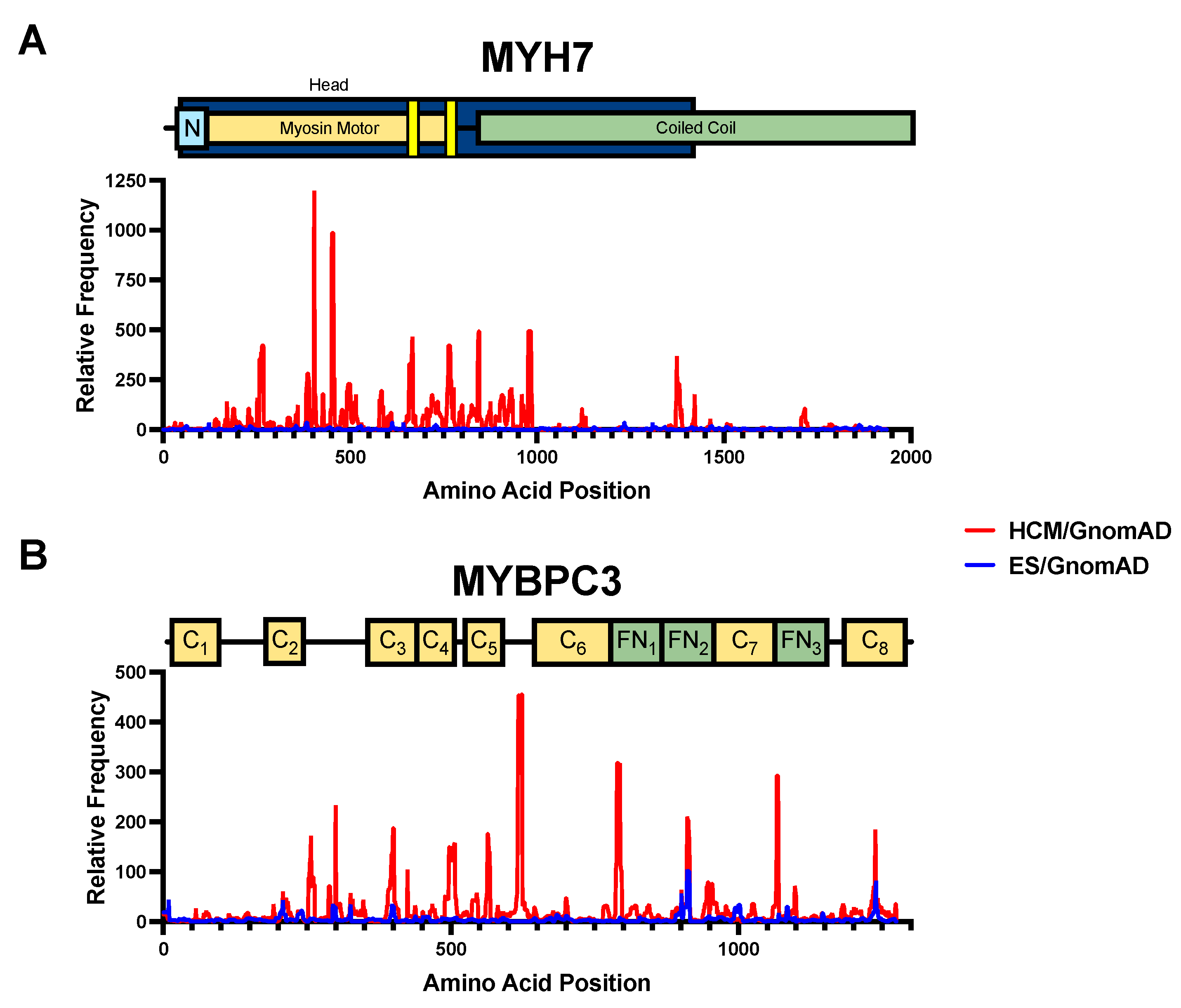
3.4. Amino Acid-Level Signal-to-Noise to Inform ACMG Pathogenicity Criteria
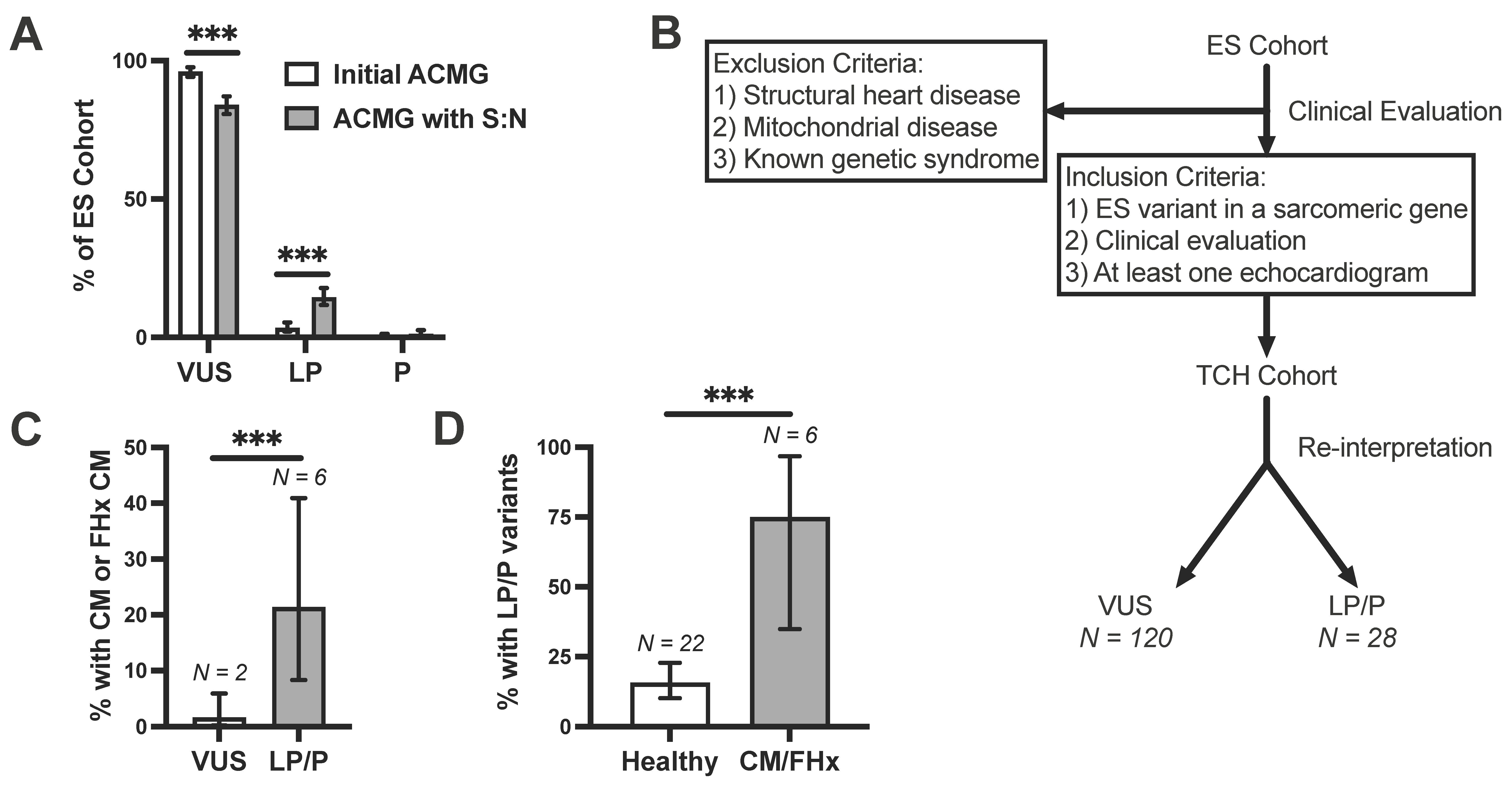
3.5. Incorporation of Signal-to-Noise Analysis into Variant Interpretation
3.6. Clinical Validation of Incidentally Identified Variants Re-Assigned as Likely Pathogenic
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| gnomAD | Genome Aggregation Database |
| HCM | hypertrophic cardiomyopathy |
| LP/P | likely pathogenic/pathogenic |
| LVH | left ventricular hypertrophy |
| LVNC | left ventricular non-compaction |
| VUS | variant of uncertain significance |
| ES | exome sequencing |
| S:N | signal-to-noise |
| ACMG | American College of Medical Genetics |
References
- Maron, B.J. Hypertrophic cardiomyopathy: A systematic review. JAMA 2002, 287, 1308–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, D.; Miltenberger-Miltenyi, G.; Vale Pereira, S.; Silva, D.; Diogo, A.N.; Madeira, H. Sarcomeric hypertrophic cardiomyopathy: Genetic profile in a Portuguese population. Rev. Port. Cardiol. 2012, 31, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Bouvagnet, P.; Chevalier, P.; Dauphin, C.; Jouk, P.S.; Da Costa, A.; Prieur, F.; Bresson, J.L.; Faivre, L.; Eicher, J.C.; et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur. J. Med. Genet. 2010, 53, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Shah, M. Hypertrophic cardiomyopathy. Cardiol. Young. 2017, 27, S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation 2019, 140, e9–e68. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e558–e631. [Google Scholar] [CrossRef]
- Authors/Task Force Members; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Thorac. Cardiovasc. Surg. 2011, 142, e153–e203. [Google Scholar] [CrossRef] [Green Version]
- Landstrom, A.P.; Kim, J.J.; Gelb, B.D.; Helm, B.M.; Kannankeril, P.J.; Semsarian, C.; Sturm, A.C.; Tristani-Firouzi, M.; Ware, S.M.; American Heart Association Council on Genomic and Precision Medicine; et al. Genetic Testing for Heritable Cardiovascular Diseases in Pediatric Patients: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2021, 14, e000086. [Google Scholar] [CrossRef]
- Musunuru, K.; Hershberger, R.E.; Day, S.M.; Klinedinst, N.J.; Landstrom, A.P.; Parikh, V.N.; Prakash, S.; Semsarian, C.; Sturm, A.C.; American Heart Association Council on Genomic and Precision Medicine; et al. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2020, 13, e000067. [Google Scholar] [CrossRef]
- Konno, T.; Chang, S.; Seidman, J.G.; Seidman, C.E. Genetics of hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2010, 25, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Bick, A.G.; Flannick, J.; Ito, K.; Cheng, S.; Vasan, R.S.; Parfenov, M.G.; Herman, D.S.; DePalma, S.R.; Gupta, N.; Gabriel, S.B.; et al. Burden of rare sarcomere gene variants in the Framingham and Jackson Heart Study cohorts. Am. J. Hum. Genet. 2012, 91, 513–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapplinger, J.D.; Landstrom, A.P.; Bos, J.M.; Salisbury, B.A.; Callis, T.E.; Ackerman, M.J. Distinguishing hypertrophic cardiomyopathy-associated mutations from background genetic noise. J. Cardiovasc. Trans. Res. 2014, 7, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Headrick, A.T.; Rosenfeld, J.A.; Yang, Y.; Tunuguntla, H.; Allen, H.D.; Penny, D.J.; Kim, J.J.; Landstrom, A.P. Incidentally identified genetic variants in arrhythmogenic right ventricular cardiomyopathy-associated genes among children undergoing exome sequencing reflect healthy population variation. Mol. Genet. Genomic. Med. 2019, 7, e593. [Google Scholar] [CrossRef] [Green Version]
- Connell, P.S.; Berkman, A.M.; Souder, B.M.; Pirozzi, E.J.; Lovin, J.J.; Rosenfeld, J.A.; Liu, P.; Tunuguntla, H.; Allen, H.D.; Denfield, S.W.; et al. Amino Acid-Level Signal-to-Noise Analysis Aids in Pathogenicity Prediction of Incidentally Identified TTN-Encoded Titin Truncating Variants. Circ. Genom. Precis. Med. 2021, 14, e003131. [Google Scholar] [CrossRef] [PubMed]
- Acmg Board of Directors. ACMG policy statement: Updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet. Med. 2015, 17, 68–69. [Google Scholar] [CrossRef] [Green Version]
- Rayment, I.; Holden, H.M.; Sellers, J.R.; Fananapazir, L.; Epstein, N.D. Structural interpretation of the mutations in the beta-cardiac myosin that have been implicated in familial hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 1995, 92, 3864–3868. [Google Scholar] [CrossRef] [Green Version]
- UniProtKB—P12883 (MYH7_HUMAN). UniProt. Available online: https://www.uniprot.org/uniprot/P12883 (accessed on 16 November 2021).
- Ezekian, J.E.; Rehder, C.; Kishnani, P.S.; Landstrom, A.P. Interpretation of Incidental Genetic Findings Localizing to Genes Associated With Cardiac Channelopathies and Cardiomyopathies. Circ. Genom. Precis. Med. 2021, 14, e003200. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landstrom, A.P.; Fernandez, E.; Rosenfeld, J.A.; Yang, Y.; Dailey-Schwartz, A.L.; Miyake, C.Y.; Allen, H.D.; Penny, D.J.; Kim, J.J. Amino acid-level signal-to-noise analysis of incidentally identified variants in genes associated with long QT syndrome during pediatric whole exome sequencing reflects background genetic noise. Heart Rhythm. 2018, 15, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Dailey-Schwartz, A.L.; Rosenfeld, J.A.; Yang, Y.; McLean, M.J.; Miyake, C.Y.; Valdes, S.O.; Fan, Y.; Allen, H.D.; Penny, D.J.; et al. Interpreting Incidentally Identified Variants in Genes Associated With Catecholaminergic Polymorphic Ventricular Tachycardia in a Large Cohort of Clinical Whole-Exome Genetic Test Referrals. Circ. Arrhythmia Electrophysiol. 2017, 10, e004742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy-A Heart Failure Society of America Practice Guideline. J. Card. Fail. 2018, 24, 281–302. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Genetics Home Reference. MYH7 Gene. U.S. National Library of Medicine. Available online: https://medlineplus.gov/genetics/gene/myh7/ (accessed on 1 July 2021).
- Richmond, C.M.; James, P.A.; Pantaleo, S.J.; Chong, B.; Lunke, S.; Tan, T.Y.; Macciocca, I. Clinical and laboratory reporting impact of ACMG-AMP and modified ClinGen variant classification frameworks in MYH7-related cardiomyopathy. Genet. Med. 2021, 23, 1108–1115. [Google Scholar] [CrossRef]
- Kelly, M.A.; Caleshu, C.; Morales, A.; Buchan, J.; Wolf, Z.; Harrison, S.M.; Cook, S.; Dillon, M.W.; Garcia, J.; Haverfield, E.; et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: Recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet. Med. 2018, 20, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Morales, A.; Kinnamon, D.D.; Jordan, E.; Platt, J.; Vatta, M.; Dorschner, M.O.; Starkey, C.A.; Mead, J.O.; Ai, T.; Burke, W.; et al. Variant Interpretation for Dilated Cardiomyopathy: Refinement of the American College of Medical Genetics and Genomics/ClinGen Guidelines for the DCM Precision Medicine Study. Circ. Genom. Precis. Med. 2020, 13, e002480. [Google Scholar] [CrossRef]
- Mademont-Soler, I.; Mates, J.; Yotti, R.; Espinosa, M.A.; Perez-Serra, A.; Fernandez-Avila, A.I.; Coll, M.; Mendez, I.; Iglesias, A.; Del Olmo, B.; et al. Additional value of screening for minor genes and copy number variants in hypertrophic cardiomyopathy. PLoS ONE 2017, 12, e0181465. [Google Scholar] [CrossRef]
- Mazzarotto, F.; Hawley, M.H.; Beltrami, M.; Beekman, L.; de Marvao, A.; McGurk, K.A.; Statton, B.; Boschi, B.; Girolami, F.; Roberts, A.M.; et al. Systematic large-scale assessment of the genetic architecture of left ventricular noncompaction reveals diverse etiologies. Genet. Med. 2021, 23, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | N (%) |
|---|---|
| Total Individuals | 7244 |
| Male | 3909 (54.0%) |
| Female | 3274 (45.2%) |
| Fetal | 61 (0.8%) |
| Age at Genetic Testing (y) | 6.1 [2.5–12.3] |
| Total Probands | 7066 |
| Unique Variants | 380 |
| LP/P | 26 (6.8% [4.3–9.4]) |
| VUS | 354 (93.2% [90.6–95.7]) |
| Variant-Positive Probands | 509 (7.2% [6.6–7.8]) |
| 1 Variant | 477 (93.7% [91.6–95.8]) |
| 2 Variants | 32 (6.3% [4.2–8.4]) |
| Probands Hosting One Variant | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Sex | Age (y) | Ethnicity | Initial Classification | Reclassification with S:N | Nucleotide | Amino Acid | Variant Type | Zygosity | Cardiac Problem | Family Cardiac Hx |
| MYH7 | F | 9.01 | African | VUS | VUS | 3301G > A | G1101S | Missense | Het | No | CM death in father at 32 |
| MYBPC3 | F | 0.06 | Caucasian | VUS | LP | 2063C > T | T688M | Missense | Het | Unspecified CM | No |
| MYBPC3 | F | 15.8 | Hispanic | VUS | VUS | 3415G > A | V1139I | Missense | Het | CM, muscular dystrophy | No |
| TPM1 | F | 1.59 | African | P | P | 475G > A | D159N | Missense | Het | LVNC | No |
| TPM1 | M | 0.54 | African | P | P | 688G > A | D230N | Missense | Het | LVNC | No |
| TNNT2 | F | 0.57 | Hispanic | LP | LP | 391C > T | R131W | Missense | Het | DCM | No |
| TNNI3 | F | 7.68 | Caucasian | VUS | LP | 400G > C | D134H | Missense | Het | DCM | No |
| Proband Hosting Two Variants | |||||||||||
| MYBPC3 | M | 6.05 | Hispanic | VUS | VUS | 2915G > A | R972Q | Missense | Het | DCM | Mother with DCM |
| MYH7 | M | LP | LP | 2710C > T | R904C | Missense | Het | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurzlechner, L.M.; Jones, E.G.; Berkman, A.M.; Tadros, H.J.; Rosenfeld, J.A.; Yang, Y.; Tunuguntla, H.; Allen, H.D.; Kim, J.J.; Landstrom, A.P. Signal-to-Noise Analysis Can Inform the Likelihood That Incidentally Identified Variants in Sarcomeric Genes Are Associated with Pediatric Cardiomyopathy. J. Pers. Med. 2022, 12, 733. https://doi.org/10.3390/jpm12050733
Kurzlechner LM, Jones EG, Berkman AM, Tadros HJ, Rosenfeld JA, Yang Y, Tunuguntla H, Allen HD, Kim JJ, Landstrom AP. Signal-to-Noise Analysis Can Inform the Likelihood That Incidentally Identified Variants in Sarcomeric Genes Are Associated with Pediatric Cardiomyopathy. Journal of Personalized Medicine. 2022; 12(5):733. https://doi.org/10.3390/jpm12050733
Chicago/Turabian StyleKurzlechner, Leonie M., Edward G. Jones, Amy M. Berkman, Hanna J. Tadros, Jill A. Rosenfeld, Yaping Yang, Hari Tunuguntla, Hugh D. Allen, Jeffrey J. Kim, and Andrew P. Landstrom. 2022. "Signal-to-Noise Analysis Can Inform the Likelihood That Incidentally Identified Variants in Sarcomeric Genes Are Associated with Pediatric Cardiomyopathy" Journal of Personalized Medicine 12, no. 5: 733. https://doi.org/10.3390/jpm12050733
APA StyleKurzlechner, L. M., Jones, E. G., Berkman, A. M., Tadros, H. J., Rosenfeld, J. A., Yang, Y., Tunuguntla, H., Allen, H. D., Kim, J. J., & Landstrom, A. P. (2022). Signal-to-Noise Analysis Can Inform the Likelihood That Incidentally Identified Variants in Sarcomeric Genes Are Associated with Pediatric Cardiomyopathy. Journal of Personalized Medicine, 12(5), 733. https://doi.org/10.3390/jpm12050733