
Targeting HIF-1α Function in Cancer through the Chaperone Action of NQO1: Implications of Genetic Diversity of NQO1

 ,
,  , , ,
, , ,  and
and 
Abstract
:1. HIF-1α: Structure, Function, Regulation and Disease
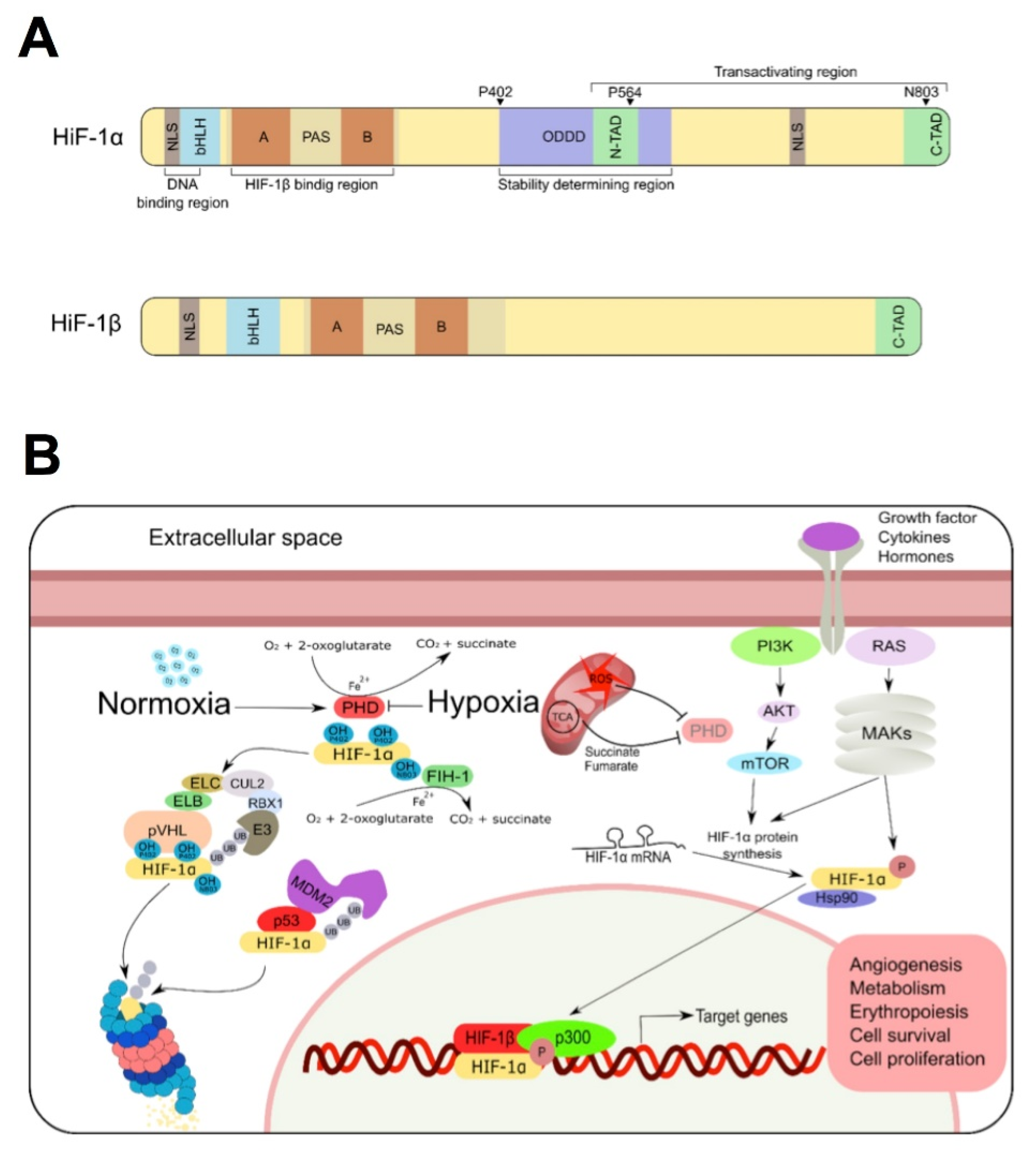
1.1. HIF-1α Structure and Function
1.2. Regulation of HIF-1α Expression
1.2.1. Oxygen-Dependent Regulation
1.2.2. Growth Factor Signaling Pathways
1.2.3. The Mdm2 Pathway
1.2.4. Heat Shock Protein 90 (Hsp90)
1.3. HIF-1α in Cancer
2. NQO1 as a Protein Chaperone: Current Knowledge and Potential Application to Target HIF-1α Stability
2.1. Overview of NQO1 Expression, Regulation and Functions: On the Potential Roles of O2 Levels and HIF-1α


2.2. NQO1 Macromolecular Interactions
2.3. Changes in NQO1 Stability, Structure and Dynamics upon Mutation and Ligand Binding: Implications for the Stability of Its Protein Partners
2.4. Mutations and Polymorphisms in NQO1: Disease and Protein Interactions
3. Targeting the Chaperone Role of NQO1 to Inactivate HIF-1α: Future Perspectives
- (i)
- To prevent PPI by targeting the protein:protein binding site or an allosteric site. We must note that allosteric communication of conformational information is well documented and likely a critical feature for the multifunctionality of both NQO1 (Section 2.3 and Section 2.4) and HIF-1α [143,144]. Allosterism in HIF-1α is dramatically exemplified by the negative effector CITED2, which competes with HIF-1α for the same binding site on CBP/p300, attenuating the transcriptional activity of HIF-1α [143,144]. Remarkably, CITED2 and HIF-1α show the same binding affinity for CBP/p300, although the former is much more efficient in displacing the latter from binary complexes with CBP/p300 due to enhanced HIF-1α release linked to the intrinsic disorder of the C-TAD domain. It is worth noting that an important caveat to rational screening for ligands is the lack of high-resolution structural models neither for a complex of NQO1 with a partner nor for the ODD of HIF-1α. There is also no detailed biochemical mapping of the interaction site or plausible molecular models. In addition to identifying non-covalent binders, potential, specific covalent modifiers should also be considered (e.g., the cysteine modification of Giardia lamblia triose phosphate isomerase by omeprazole which destabilizes the dimer [145]);
- (ii)
- A high-throughput screening for ligands that targets the formation of the NQO1:HIF-1α complex. Both (i) and (ii) would require a rapid, reproducible, robust assay for the interaction in vitro, for example labeling of the proteins with a fluorophore and a quencher. A cell-based assay would also be required to test hits from the screen under in vivo conditions, for example proteins labeled with FRET donors and acceptors, co-immunoprecipitation or indirectly by measuring amounts of HIF1α. Although challenging due to several highly disordered regions in the HIF-1α protein, such screening would still be feasible as recently reported for inhibiting PPI in the cancer-associated and intrinsically disordered protein NUPR1 [146,147];
- (iii)
- To use dicoumarol-like molecules that may target that interaction with lower second-site effects. It must be noted regarding this approach that the effects of NADH or dicoumarol have not been tested for the interaction of NQO1 with HIF-1α. Many dicoumarol analogues that function as NQO1 inhibitors have been reported [148,149,150]. It is reasonable to assume that they would also antagonize the NQO1 interactions with binding partners in a similar way to dicoumarol. As such, they represent immediately available compounds that could be tested for their ability to antagonize the NQO1:HIF1α interaction. However, they are likely to inhibit or antagonize many of NQO1′s functions and may, therefore, cause significant off-target effects. Some of these may be undesirable in the context of cancer therapy, e.g., the antagonism of the NQO1/p53 interaction and consequent down-regulation of p53-mediated apoptosis [77,151].
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Comments | References |
|---|---|---|
| Substrates | ||
| Dichlorophenolindolphenol (DCPIP) | Non-physiological, but often used in in vitro assays | [152,153] |
| Menadione (Vitamin K3) | Reaction occurs in vivo but enzyme likely to play only a minor role in blood clotting | [72,153] |
| Coenzyme Q10 (ubiquinone) | NQO1 maintains this, and related compounds, in the reduced form | [154,155] |
| Superoxide ion | Likely physiological role in directly combatting oxidative stress in vivo. | [75] |
| Fe(III) ion | Probably non-physiological | [156] |
| Idebenone | Important in the metabolism of this drug (a coenzyme Q10 mimic) | [157] |
| (3-hydroxymethyl-5-aziridinyl-1-methyl-2-(H-indole-4, 7-indione)-propenol) EO9 | Reduction activates this anticancer compound | [158] |
| Quinone epoxides | Potentially important if members of this group of compounds used as drugs. | [159] |
| β-lapachone | Futile cycling involving NQO1 results in reduced cellular concentrations of NAD(P)H contributing to cell death | [160,161] |
| Mitomycin C | Reduction activates this akylating cytotoxic drug | [162,163] |
| Tirapazamine and other heteroaromatic N-oxides | Slow reaction. Superoxide also produced | [164] |
| Benzofuroxans | Reduction by NQO1 may play a minor role in cytotoxicity | [165] |
| Nitroaromatics | Reactivity correlates with electrode potential | [166,167] |
| Aminochrome | Reaction is important in protection against Parkinson’s Disease and other neurological diseases | [168,169] |
| Inhibitors | ||
| Dicoumarol and derivatives thereof | High affinity; competes with NAD(P)H; negatively cooperative; often used in experimental studies; derivatives may be anticancer lead compounds; dissociates NQO1-p53 complexes resulting in increased p53 degradation and inhibition of apoptosis | [77,141,149,153,170,171] |
| Curcumin | May dissociate NQO1-p53 complexes resulting in increased p53 degradation and inhibition of apoptosis. Other studies suggest it may enhance the NQO1-p53 interaction in vivo | [172,173] |
| Resveratrol | Potent inhibitor of the related protein NQO2; only weakly inhibits NQO1 | [174] |
| Warfarin | NQO1 is a secondary target for this anticoagulant | [171,175] |
| Macromolecule | Effect on Partner Stability | Effect of NAD(P)H | Effect of Dicoumarol | General Comments | References |
|---|---|---|---|---|---|
| p53 | Binds to, and stabilizes, the full-length protein, protecting it from proteasomal degradation | Increases affinity of interaction. | Antagonizes interaction. | Interaction promotes p53-mediated apoptosis. Dicoumarol down-regulates this by promoting release of p53 from NQO1 and consequent degradation of p53. | [84,141,151,176,177] |
| p73α | Binds to, and stabilizes, the full-length protein, protecting it from proteasomal degradation. | Increases affinity of interaction; effect not observed with NAD+. | Antagonizes interaction. | Interaction promotes p73α-mediated apoptosis. Dicoumarol down-regulates this by promoting release of p73α from NQO1 and consequent degradation of p73α. No interaction with p73β which lacks a SAM domain at the C-terminus. The SAM in p73α is responsible for the interaction. | [67,84] |
| Ornithine decarboxylase (ODC) | NQO1 binds to, and stabilizes ODC, preventing proteasomal degradation | Not known. | Antagonizes interaction | ODC monomer (inactive) degradation is enhanced by binding antizyme 1 (AZ1) which targets the ODC/AZ1 complex to the proteasome. NQO1 protects monomeric ODC by binding the ODC/AZ1 heterodimer. | [114,178] |
| mRNA encoding SERPINA1 (α-1-antitrypsin) | No effect on stability | Not known | Not known | Does not affect the amount of mRNA, but does enhance the translation by binding to 3′-UTR. This results in more protein | [113] |
| 20S proteasomal subunit | Not known | No effect | Not known | NQO1 interacts with the proteasome and negatively regulates proteolytic activity. NQO1-apo is degraded by the proteasome. | [116] |
| HIF-1α | NQO1 binds HIF-1α, stabilizes it and prevent proteasomal degradation | Not known | Not known | Interaction occurs in cytoplasm. NQO1 enhances transcription of HIF-1α regulated genes, presumably by increasing the amount of HIF-1α. See also main text. | [63] |
| Hsp70/HSPA4 | Interaction most likely occurs with newly synthesized NQO1, presumably stabilizing it and assisting folding. | Not known | Not known | NQO1-p.P187S only interacts very weakly. | [179] |
| STUB1/CHIP | Not known | Not known | Not known | NQO1 ubiquitination is mediated upon binding to STUB1 which triggers NQO1 degradation. NQO1-p.P187S and apo-NQO1 are degraded efficiently by this mechanism. STUB1′s TPR domain is required for the interaction. | [117,180] |
| c-FOS | NQO1 binding to c-FOS protects the former from 20S proteasomal degradation | Not known | Not known | NQO1 localizes c-Fos (at least partly) to the cytoplasm. Free c-Fos or c-Fos in complex with other transcription factors, is localized to the nucleus. | [181] |
| Bcl2-associated Athanogene 3 (BAG3) | Not known | Not known | Not known | BAG3 regulates the proteasome. siRNA knockdown of BAG3 reduces proteasomal activity. | [182] |
| ING1B (p33) | NQO1 binds to p33ING1b tumor suppressor protecting the former from 20S proteasomal degradation. | Increases affinity of interaction | Not known | NQO1 binds preferentially to Ser-126 phosphorylated p33ING1b. This phosphorylation is induced by genotoxoic stress and increases the in vivo half-life of the protein from 5.7 h to 16.8 h. | [183,184] |
| HIV-1 Tat | HIV-1 Tat binds to NQO1 stabilizing the former | Not known, but assumed to promote interaction. | Antagonizes interaction. | HIV Rev downregulates NQO1 destabilizing HIV/Tat complex and thus resulting in increased Tat degradation. Dicoumarol inhibits HIV replication. | [135] |
| eIF4GI | eIF4GI binds to NQO1 leading to protection of the former against proteasomal degradation | Not known. | Antagonizes interaction. | This interaction modulates mRNA translation. Dicoumarol downregulates translation. Interaction does not require RNA binding by eIF4GI. Oxidative stress reduces the amount of cellular eIF4GI. | [185] |
| Homocysteine-induced endoplasmic reticulum protein (Herp) | Stabilizes Herp by protecting it from proteasomal degradation | Not known | Antagonizes interaction | Herp is up-regulated in the unfolded protein response (UPR). Herp’s cellular half-life is increased by NQO1. | [186,187] |
| PGC-1α | Stabilizes this intrinsically disordered protein and protects it from proteasomal degradation. | Enhances interaction | Antagonizes interaction | Cellular levels of NQO1 and PGC-1α are correlated. Stabilization of PGC-1α by NQO1 leads to induction of genes encoding enzymes of gluconeogénesis. | [188] |
| RIL (reversion-induced LIM domain protein; PDLIM4) | NQO1 binds and stabilizes unstructured C-terminal region of one alternately spliced isoform (RILaltCterm) protecting it from proteasomal degradation. | Not known | Increases RIL proteasomal degradation, presumably by antagonizing RIL/NQO1 interaction | RILaltCterm accumulates in response to oxidative stress and stimulates actin cytoskeleton rearrangement. | [189] |
| TAp63γ | Stabilizes TAp63γ and protects it from proteasomal degradation. | Not known | Not known | NQO1-p.P187S does not interact. Interaction occurs in response to genotoxic stress. | [190] |
References
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, M.S.; Keith, B.; Simon, M.C. Oxygen availability and metabolic adaptations. Nat. Rev. Cancer 2016, 16, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF-1α and HIF-2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee Koh, M.; Spivak-Kroizman, T.R.; Powis, G. HIF-1 regulation: Not so easy come, easy go. Trends Biochem. Sci. 2008, 33, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef] [Green Version]
- Makino, Y.; Cao, R.; Svensson, K.; Bertilsson, G.; Asman, M.; Tanaka, H.; Cao, Y.; Berkenstam, A.; Poellinger, L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 2001, 414, 550–554. [Google Scholar] [CrossRef]
- Marti, H.H.; Katschinski, D.M.; Wagner, K.F.; Schaffer, L.; Stier, B.; Wenger, R.H. Isoform-specific expression of hypoxia-inducible factor-1alpha during the late stages of mouse spermiogenesis. Mol. Endocrinol. 2002, 16, 234–243. [Google Scholar]
- Maynard, M.A.; Qi, H.; Chung, J.; Lee, E.H.; Kondo, Y.; Hara, S.; Conaway, R.C.; Conaway, J.W.; Ohh, M. Multiple splice variants of the human HIF-3 alpha locus are targets of the von Hippel-Lindau E3 ubiquitin ligase complex. J. Biol. Chem. 2003, 278, 11032–11040. [Google Scholar] [CrossRef] [Green Version]
- Depping, R.; Hagele, S.; Wagner, K.F.; Wiesner, R.J.; Camenisch, G.; Wenger, R.H.; Katschinski, D.M. A dominant-negative isoform of hypoxia-inducible factor-1 alpha specifically expressed in human testis. Biol. Reprod. 2004, 71, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallio, P.J.; Okamoto, K.; O’Brien, S.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998, 17, 6573–6586. [Google Scholar] [CrossRef] [PubMed]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, K.; Discher, D.J.; Hu, J.; Bishopric, N.H.; Webster, K.A. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J. Biol. Chem. 2001, 276, 12645–12653. [Google Scholar] [CrossRef] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamura, T.; Sato, S.; Iwai, K.; Czyzyk-Krzeska, M.; Conaway, R.C.; Conaway, J.W. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc. Natl. Acad. Sci. USA 2000, 97, 10430–10435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [Green Version]
- BelAiba, R.S.; Bonello, S.; Zähringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Görlach, A. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol. Biol. Cell 2007, 18, 4691–4697. [Google Scholar] [CrossRef] [Green Version]
- El-Naggar, A.M.; Sorensen, P.H. Translational control of aberrant stress responses as a hallmark of cancer. J. Pathol. 2018, 244, 650–666. [Google Scholar] [CrossRef] [Green Version]
- Hagen, T.; Taylor, C.T.; Lam, F.; Moncada, S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: Effect on HIF1alpha. Science 2003, 302, 1975–1978. [Google Scholar] [CrossRef]
- Doege, K.; Heine, S.; Jensen, I.; Jelkmann, W.; Metzen, E. Inhibition of mitochondrial respiration elevates oxygen concentration but leaves regulation of hypoxia-inducible factor (HIF) intact. Blood 2005, 106, 2311–2317. [Google Scholar] [CrossRef] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Agani, F.H.; Pichiule, P.; Chavez, J.C.; LaManna, J.C. The role of mitochondria in the regulation of hypoxia-inducible factor 1 expression during hypoxia. J. Biol. Chem. 2000, 275, 35863–35867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalgard, C.L.; Lu, H.; Mohyeldin, A.; Verma, A. Endogenous 2-oxoacids differentially regulate expression of oxygen sensors. Biochem. J. 2004, 380, 419–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Koivunen, P.; Hirsila, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [Green Version]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Feldser, D.; Agani, F.; Iyer, N.V.; Pak, B.; Ferreira, G.; Semenza, G.L. Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res. 1999, 59, 3915–3918. [Google Scholar]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [Green Version]
- Zelzer, E.; Levy, Y.; Kahana, C.; Shilo, B.Z.; Rubinstein, M.; Cohen, B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO J. 1998, 17, 5085–5094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.; Isaacs, J.S.; Lee, S.; Trepel, J.; Liu, Z.G.; Neckers, L. Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochem. J. 2003, 370, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Isaacs, J.S.; Lee, S.; Trepel, J.; Neckers, L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003, 17, 2115–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albina, J.E.; Mastrofrancesco, B.; Vessella, J.A.; Louis, C.A.; Henry, W.L., Jr.; Reichner, J.S. HIF-1 expression in healing wounds: HIF-1alpha induction in primary inflammatory cells by TNF-alpha. Am. J. Physiol. Cell Physiol. 2001, 281, C1971–C1977. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Sonenberg, N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001, 15, 807–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, N.; Stiehl, D.P.; Bohensky, J.; Leshchinsky, I.; Srinivas, V.; Caro, J. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J. Biol. Chem. 2003, 278, 14013–14019. [Google Scholar] [CrossRef] [Green Version]
- Sonenberg, N.; Hinnebusch, A.G. New modes of translational control in development, behavior, and disease. Mol. Cell 2007, 28, 721–729. [Google Scholar] [CrossRef]
- Richard, D.E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Li, M.; Luo, J.; Gu, W. Direct interactions between HIF-1 alpha and Mdm2 modulate p53 function. J. Biol. Chem. 2003, 278, 13595–13598. [Google Scholar] [CrossRef] [Green Version]
- Schmid, T.; Zhou, J.; Kohl, R.; Brune, B. p300 relieves p53-evoked transcriptional repression of hypoxia-inducible factor-1 (HIF-1). Biochem. J. 2004, 380, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Jung, Y.J.; Mimnaugh, E.G.; Martinez, A.; Cuttitta, F.; Neckers, L.M. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J. Biol. Chem. 2002, 277, 29936–29944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Lin, Z.; Liang, D.; Fath, D.; Sang, N.; Caro, J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol. Cell. Biol. 2006, 26, 2019–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradin, K.; McGuire, J.; Wenger, R.H.; Kvietikova, I.; Fhitelaw, M.L.; Toftgard, R.; Tora, L.; Gassmann, M.; Poellinger, L. Functional interference between hypoxia and dioxin signal transduction pathways: Competition for recruitment of the Arnt transcription factor. Mol. Cell. Biol. 1996, 16, 5221–5231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Thomlinson, R.H.; Gray, L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Moulder, J.E.; Rockwell, S. Hypoxic fractions of solid tumors: Experimental techniques, methods of analysis, and a survey of existing data. Int. J. Radiat. Oncol. Biol. Phys. 1984, 10, 695–712. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Jiang, H. Prediction of postoperative survival of triple-negative breast cancer based on nomogram model combined with expression of HIF-1alpha and c-myc. Medicine 2019, 98, e17370. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.T.; Kim, J.W.; Kim, J.M.; Kim, S.J.; Lee, J.S.; Hong, S.S.; Goodwin, J.; Ruthenborg, R.J.; Jung, M.G.; Lee, H.J.; et al. NQO1 inhibits proteasome-mediated degradation of HIF-1alpha. Nat. Commun. 2016, 7, 13593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Bianchet, M.A.; Talalay, P.; Amzel, L.M. The three-dimensional structure of NAD(P)H:quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: Mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. USA 1995, 92, 8846–8850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaver, S.K.; Mesa-Torres, N.; Pey, A.L.; Timson, D.J. NQO1: A target for the treatment of cancer and neurological diseases, and a model to understand loss of function disease mechanisms. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Megarity, C.F.; Medina-Carmona, E.; Timson, D.J. Natural Small Molecules as Stabilizers and Activators of Cancer-Associated NQO1 Polymorphisms. Curr. Drug Targets 2016, 17, 1506–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Carmona, E.; Neira, J.L.; Salido, E.; Fuchs, J.E.; Palomino-Morales, R.; Timson, D.J.; Pey, A.L. Site-to-site interdomain communication may mediate different loss-of-function mechanisms in a cancer-associated NQO1 polymorphism. Sci. Rep. 2017, 7, 44352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lienhart, W.D.; Gudipati, V.; Uhl, M.K.; Binter, A.; Pulido, S.A.; Saf, R.; Zangger, K.; Gruber, K.; Macheroux, P. Collapse of the native structure caused by a single amino acid exchange in human NAD(P)H:quinone oxidoreductase(1.). FEBS J. 2014, 281, 4691–4704. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Deng, P.S.; Bailey, J.M.; Swiderek, K.M. A two-domain structure for the two subunits of NAD(P)H:quinone acceptor oxidoreductase. Protein Sci. 1994, 3, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Faig, M.; Bianchet, M.A.; Talalay, P.; Chen, S.; Winski, S.; Ross, D.; Amzel, L.M. Structures of recombinant human and mouse NAD(P)H:quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. USA 2000, 97, 3177–3182. [Google Scholar] [CrossRef]
- Anusevicius, Z.; Sarlauskas, J.; Cenas, N. Two-electron reduction of quinones by rat liver NAD(P)H:quinone oxidoreductase: Quantitative structure-activity relationships. Arch. Biochem. Biophys. 2002, 404, 254–262. [Google Scholar] [CrossRef]
- Ingram, B.O.; Turbyfill, J.L.; Bledsoe, P.J.; Jaiswal, A.K.; Stafford, D.W. Assessment of the contribution of NAD(P)H-dependent quinone oxidoreductase 1 (NQO1) to the reduction of vitamin K in wild-type and NQO1-deficient mice. Biochem. J. 2013, 456, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landi, L.; Fiorentini, D.; Galli, M.C.; Segura-Aguilar, J.; Beyer, R.E. DT-Diaphorase maintains the reduced state of ubiquinones in lipid vesicles thereby promoting their antioxidant function. Free Radic. Biol. Med. 1997, 22, 329–335. [Google Scholar] [CrossRef]
- Siegel, D.; Bolton, E.M.; Burr, J.A.; Liebler, D.C.; Ross, D. The reduction of alpha-tocopherolquinone by human NAD(P)H: Quinone oxidoreductase: The role of alpha-tocopherolhydroquinone as a cellular antioxidant. Mol. Pharmacol. 1997, 52, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.; Gustafson, D.L.; Dehn, D.L.; Han, J.Y.; Boonchoong, P.; Berliner, L.J.; Ross, D. NAD(P)H:quinone oxidoreductase 1: Role as a superoxide scavenger. Mol. Pharmacol. 2004, 65, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Anoz-Carbonell, E.; Timson, D.J.; Pey, A.L.; Medina, M. The Catalytic Cycle of the Antioxidant and Cancer-Associated Human NQO1 Enzyme: Hydride Transfer, Conformational Dynamics and Functional Cooperativity. Antioxidants 2020, 9, 772. [Google Scholar] [CrossRef]
- Megarity, C.F.; Abdel-Bettley, H.; Caraher, M.C.; Scott, K.A.; Jowitt, T.A.; Gutierrez, A.; Bryce, R.A.; Nolan, K.A.; Stratford, I.J.; Timson, D.; et al. Negative cooperativity in NADP(H) quinone oxidoreductase 1 (NQO1). ChemBioChem 2019, 20, 2841–2849. [Google Scholar] [CrossRef]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. FAD binding overcomes defects in activity and stability displayed by cancer-associated variants of human NQO1. Biochim. Biophys. Acta 2014, 1842, 2163–2173. [Google Scholar] [CrossRef] [Green Version]
- Claveria-Gimeno, R.; Velazquez-Campoy, A.; Pey, A.L. Thermodynamics of cooperative binding of FAD to human NQO1: Implications to understanding cofactor-dependent function and stability of the flavoproteome. Arch. Biochem. Biophys. 2017, 636, 17–27. [Google Scholar] [CrossRef]
- Timson, D.J. Dicoumarol: A Drug which Hits at Least Two Very Different Targets in Vitamin K Metabolism. Curr. Drug Targets 2017, 18, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Cullen, J.J.; Hinkhouse, M.M.; Grady, M.; Gaut, A.W.; Liu, J.; Zhang, Y.P.; Weydert, C.J.; Domann, F.E.; Oberley, L.W. Dicumarol inhibition of NADPH:quinone oxidoreductase induces growth inhibition of pancreatic cancer via a superoxide-mediated mechanism. Cancer Res. 2003, 63, 5513–5520. [Google Scholar] [PubMed]
- Lee, H.; Oh, E.T.; Choi, B.H.; Park, M.T.; Lee, J.K.; Lee, J.S.; Park, H.J. NQO1-induced activation of AMPK contributes to cancer cell death by oxygen-glucose deprivation. Sci. Rep. 2015, 5, 7769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The crystal structure of NAD(P)H quinone oxidoreductase 1 in complex with its potent inhibitor dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.; Dehn, D.D.; Bokatzian, S.S.; Quinn, K.; Backos, D.S.; Di Francesco, A.; Bernier, M.; Reisdorph, N.; de Cabo, R.; Ross, D. Redox modulation of NQO1. PLoS ONE 2018, 13, e0190717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.; Bersie, S.; Harris, P.; Di Francesco, A.; Armstrong, M.; Reisdorph, N.; Bernier, M.; de Cabo, R.; Fritz, K.; Ross, D. A redox-mediated conformational change in NQO1 controls binding to microtubules and α-tubulin acetylation. Redox Biol. 2021, 39, 101840. [Google Scholar] [CrossRef]
- Luo, S.; Su Kang, S.; Wang, Z.H.; Liu, X.; Day, J.X.; Wu, Z.; Peng, J.; Xiang, D.; Springer, W.; Ye, K. Akt Phosphorylates NQO1 and Triggers its Degradation, Abolishing its Antioxidative Activities in Parkinson’s Disease. J. Neurosci. 2019, 39, 7291–7305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Carmona, E.; Rizzuti, B.; Martin-Escolano, R.; Pacheco-Garcia, J.L.; Mesa-Torres, N.; Neira, J.L.; Guzzi, R.; Pey, A.L. Phosphorylation compromises FAD binding and intracellular stability of wild-type and cancer-associated NQO1: Insights into flavo-proteome stability. Int. J. Biol. Macromol. 2019, 125, 1275–1288. [Google Scholar] [CrossRef]
- Ross, D.; Kepa, J.K.; Winski, S.L.; Beall, H.D.; Anwar, A.; Siegel, D. NAD(P)H:quinone oxidoreductase 1 (NQO1): Chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem. Biol. Interact. 2000, 129, 77–97. [Google Scholar] [CrossRef]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [CrossRef]
- Nioi, P.; Hayes, J.D. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat. Res. 2004, 555, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Brauze, D.; Widerak, M.; Cwykiel, J.; Szyfter, K.; Baer-Dubowska, W. The effect of aryl hydrocarbon receptor ligands on the expression of AhR, AhRR, ARNT, Hif1alpha, CYP1A1 and NQO1 genes in rat liver. Toxicol. Lett. 2006, 167, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, H.; O’Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 2002, 294, 1–12. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [Green Version]
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The Mediator Subunit MED16 Transduces NRF2-Activating Signals into Antioxidant Gene Expression. Mol. Cell. Biol. 2016, 36, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valerio, L.G., Jr.; Kepa, J.K.; Pickwell, G.V.; Quattrochi, L.C. Induction of human NAD(P)H:quinone oxidoreductase (NQO1) gene expression by the flavonol quercetin. Toxicol. Lett. 2001, 119, 49–57. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Lu, X.; Wang, Z.; Wu, J.M. Induction of quinone reductase NQO1 by resveratrol in human K562 cells involves the antioxidant response element ARE and is accompanied by nuclear translocation of transcription factor Nrf2. Med. Chem. 2006, 2, 275–285. [Google Scholar] [PubMed]
- Waleh, N.S.; Calaoagan, J.; Murphy, B.J.; Knapp, A.M.; Sutherland, R.M.; Laderoute, K.R. The redox-sensitive human antioxidant responsive element induces gene expression under low oxygen conditions. Carcinogenesis 1998, 19, 1333–1337. [Google Scholar] [CrossRef] [Green Version]
- O’Dwyer, P.J.; Yao, K.S.; Ford, P.; Godwin, A.K.; Clayton, M. Effects of hypoxia on detoxicating enzyme activity and expression in HT29 colon adenocarcinoma cells. Cancer Res. 1994, 54, 3082–3087. [Google Scholar]
- Beischlag, T.V.; Luis Morales, J.; Hollingshead, B.D.; Perdew, G.H. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 207–250. [Google Scholar] [CrossRef] [Green Version]
- Rowlands, J.C.; Gustafsson, J.A. Aryl hydrocarbon receptor-mediated signal transduction. Crit. Rev. Toxicol. 1997, 27, 109–134. [Google Scholar] [CrossRef]
- Ma, Q.; Kinneer, K.; Bi, Y.; Chan, J.Y.; Kan, Y.W. Induction of murine NAD(P)H:quinone oxidoreductase by 2,3,7,8-tetrachlorodibenzo-p-dioxin requires the CNC (cap ‘n’ collar) basic leucine zipper transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2): Cross-interaction between AhR (aryl hydrocarbon receptor) and Nrf2 signal transduction. Biochem. J. 2004, 377, 205–213. [Google Scholar]
- Gassmann, M.; Kvietikova, I.; Rolfs, A.; Wenger, R.H. Oxygen- and dioxin-regulated gene expression in mouse hepatoma cells. Kidney Int. 1997, 51, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Yeligar, S.M.; Machida, K.; Kalra, V.K. Ethanol-induced HO-1 and NQO1 are differentially regulated by HIF-1alpha and Nrf2 to attenuate inflammatory cytokine expression. J. Biol. Chem. 2010, 285, 35359–35373. [Google Scholar] [CrossRef] [Green Version]
- Medina-Carmona, E.; Palomino-Morales, R.J.; Fuchs, J.E.; Padín-Gonzalez, E.; Mesa-Torres, N.; Salido, E.; Timson, D.J.; Pey, A.L. Conformational dynamics is key to understanding loss-of-function of NQO1 cancer-associated polymorphisms and its correction by pharmacological ligands. Sci. Rep. 2016, 6, 20331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Francesco, A.; Di Germanio, C.; Panda, A.C.; Huynh, P.; Peaden, R.; Navas-Enamorado, I.; Bastian, P.; Lehrmann, E.; Diaz-Ruiz, A.; Ross, D.; et al. Novel RNA-binding activity of NQO1 promotes SERPINA1 mRNA translation. Free Radic. Biol. Med. 2016, 99, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Asher, G.; Bercovich, Z.; Tsvetkov, P.; Shaul, Y.; Kahana, C. 20S proteasomal degradation of ornithine decarboxylase is regulated by NQO1. Mol. Cell 2005, 17, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscovitz, O.; Tsvetkov, P.; Hazan, N.; Michaelevski, I.; Keisar, H.; Ben-Nissan, G.; Shaul, Y.; Sharon, M. A mutually inhibitory feedback loop between the 20S proteasome and its regulator, NQO1. Mol. Cell 2012, 47, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Limon, A.; Alriquet, M.; Lang, W.H.; Calloni, G.; Wittig, I.; Vabulas, R.M. Recognition of enzymes lacking bound cofactor by protein quality control. Proc. Natl. Acad. Sci. USA 2016, 113, 12156–12161. [Google Scholar] [CrossRef] [Green Version]
- Medina-Carmona, E.; Fuchs, J.E.; Gavira, J.A.; Mesa-Torres, N.; Neira, J.L.; Salido, E.; Palomino-Morales, R.; Burgos, M.; Timson, D.J.; Pey, A.L. Enhanced vulnerability of human proteins towards disease-associated inactivation through divergent evolution. Hum. Mol. Genet. 2017, 26, 3531–3544. [Google Scholar] [CrossRef]
- Pey, A.L. Anion-specific interaction with human NQO1 inhibits flavin binding. Int. J. Biol. Macromol. 2019, 126, 1223–1233. [Google Scholar] [CrossRef]
- Pey, A.L. Biophysical and functional perturbation analyses at cancer-associated P187 and K240 sites of the multifunctional NADP(H):quinone oxidoreductase 1. Int. J. Biol. Macromol. 2018, 118, 1912–1923. [Google Scholar] [CrossRef]
- Siegel, D.; Anwar, A.; Winski, S.L.; Kepa, J.K.; Zolman, K.L.; Ross, D. Rapid polyubiquitination and proteasomal degradation of a mutant form of NAD(P)H:quinone oxidoreductase 1. Mol. Pharmacol. 2001, 59, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Ruiz, A.; Lanasa, M.; Garcia, J.; Mora, H.; Fan, F.; Martin-Montalvo, A.; Di Francesco, A.; Calvo-Rubio, M.; Salvador-Pascual, A.; Aon, M.A.; et al. Overexpression of CYB5R3 and NQO1, two NAD(+)-producing enzymes, mimics aspects of caloric restriction. Aging Cell 2018, 17, e12767. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and Functions of Spatial Protein Quality Control. Annu. Rev. Biochem. 2017, 86, 97–122. [Google Scholar] [CrossRef] [PubMed]
- Enam, C.; Geffen, Y.; Ravid, T.; Gardner, R.G. Protein Quality Control Degradation in the Nucleus. Annu. Rev. Biochem. 2018, 87, 725–749. [Google Scholar] [CrossRef]
- Fang, Q.; Andrews, J.; Sharma, N.; Wilk, A.; Clark, J.; Slyskova, J.; Koczor, C.A.; Lans, H.; Prakash, A.; Sobol, R.W. Stability and sub-cellular localization of DNA polymerase beta is regulated by interactions with NQO1 and XRCC1 in response to oxidative stress. Nucleic Acids Res. 2019, 47, 6269–6286. [Google Scholar] [CrossRef] [Green Version]
- Janke, C.; Montagnac, G. Causes and Consequences of Microtubule Acetylation. Curr. Biol. 2017, 27, R1287–R1292. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Lienhart, W.D.; Strandback, E.; Gudipati, V.; Koch, K.; Binter, A.; Uhl, M.K.; Rantasa, D.M.; Bourgeois, B.; Madl, T.; Zangger, K.; et al. Catalytic competence, structure and stability of the cancer-associated R139W variant of the human NAD(P)H:quinone oxidoreductase 1 (NQO1). FEBS J. 2017, 284, 1233–1245. [Google Scholar] [CrossRef] [Green Version]
- Munoz, I.G.; Morel, B.; Medina-Carmona, E.; Pey, A.L. A mechanism for cancer-associated inactivation of NQO1 due to P187S and its reactivation by the consensus mutation H80R. FEBS Lett. 2017, 591, 2826–2835. [Google Scholar] [CrossRef] [Green Version]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. NAD(P)H quinone oxidoreductase (NQO1): An enzyme which needs just enough mobility, in just the right places. Biosci. Rep. 2019, 39, BSR20180459. [Google Scholar] [CrossRef] [Green Version]
- Pacheco-Garcia, J.L.; Anoz-Carbonell, E.; Vankova, P.; Kannan, A.; Palomino-Morales, R.; Mesa-Torres, N.; Salido, E.; Man, P.; Medina, M.; Naganathan, A.N.; et al. Structural basis of the pleiotropic and specific phenotypic consequences of missense mutations in the multifunctional NAD(P)H:quinone oxidoreductase 1 and their pharmacological rescue. Redox Biol. 2021, 46, 102112. [Google Scholar] [CrossRef] [PubMed]
- Vankova, P.; Salido, E.; Timson, D.J.; Man, P.; Pey, A.L. A dynamic core in human NQO1 controls the functional and stability effects of ligand binding and their communication across the enzyme dimer. Biomolecules 2019, 9, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Carmona, E.; Betancor-Fernández, I.; Santos, J.; Mesa-Torres, N.; Grottelli, S.; Batlle, C.; Naganathan, A.N.; Oppici, O.; Cellini, B.; Ventura, S.; et al. Insight into the specificity and severity of pathogenic mechanisms associated with missense mutations through experimental and structural perturbation analyses. Hum. Mol. Genet. 2019, 28, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mesa-Torres, N.; Betancor-Fernández, I.; Oppici, E.; Cellini, B.; Salido, E.; Pey, A.L. Evolutionary Divergent Suppressor Mutations in Conformational Diseases. Genes 2018, 9, 352. [Google Scholar] [CrossRef] [Green Version]
- Lata, S.; Ali, A.; Sood, V.; Raja, R.; Banerjea, A.C. HIV-1 Rev downregulates Tat expression and viral replication via modulation of NAD(P)H:quinine oxidoreductase 1 (NQO1). Nat. Commun. 2015, 6, 7244. [Google Scholar] [CrossRef]
- Moran, J.L.; Siegel, D.; Ross, D. A potential mechanism underlying the increased susceptibility of individuals with a polymorphism in NAD(P)H:quinone oxidoreductase 1 (NQO1) to benzene toxicity. Proc. Natl. Acad. Sci. USA 1999, 96, 8150–8155. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.; McGuinness, S.M.; Winski, S.L.; Ross, D. Genotype-phenotype relationships in studies of a polymorphism in NAD(P)H:quinone oxidoreductase 1. Pharmacogenetics 1999, 9, 113–121. [Google Scholar] [CrossRef]
- Traver, R.D.; Siegel, D.; Beall, H.D.; Phillips, R.M.; Gibson, N.W.; Franklin, W.A.; Ross, D. Characterization of a polymorphism in NAD(P)H: Quinone oxidoreductase (DT-diaphorase). Br. J. Cancer 1997, 75, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.S.; Forrest, G.L.; Akman, S.A.; Hu, L.T. NAD(P)H:quinone oxidoreductase expression and mitomycin C resistance developed by human colon cancer HCT 116 cells. Cancer Res. 1995, 55, 330–335. [Google Scholar]
- Pacheco-Garciía, J.L.; Cano-Munñoz, M.; Saánchez-Ramos, I.; Salido, E.; Pey, A.L. Naturally-Occurring Rare Mutations Cause Mild to Catastrophic Effects in the Multifunctional and Cancer-Associated NQO1 Protein. J. Pers. Med. 2020, 10, 207. [Google Scholar] [CrossRef]
- Asher, G.; Lotem, J.; Kama, R.; Sachs, L.; Shaul, Y. NQO1 stabilizes p53 through a distinct pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 3099–3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Perri, J.I.; Dengler, V.L.; Audetat, K.A.; Pandey, A.; Bonner, E.A.; Urh, M.; Mendez, J.; Daniels, D.L.; Wappner, P.; Galbraith, M.D.; et al. The TIP60 Complex Is a Conserved Coactivator of HIF1A. Cell Rep. 2016, 16, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature 2017, 543, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Brooks Iii, C.L. Electrostatic Forces Control the Negative Allosteric Regulation in a Disordered Protein Switch. J. Phys. Chem. Lett. 2020, 11, 864–868. [Google Scholar] [CrossRef]
- Garcia-Torres, I.; de la Mora-de la Mora, I.; Marcial-Quino, J.; Gomez-Manzo, S.; Vanoye-Carlo, A.; Navarrete-Vazquez, G.; Colin-Lozano, B.; Gutierrez-Castrellon, P.; Sierra-Palacios, E.; Lopez-Velazquez, G.; et al. Proton pump inhibitors drastically modify triosephosphate isomerase from Giardia lamblia at functional and structural levels, providing molecular leads in the design of new antigiardiasic drugs. Biochim. Biophys. Acta 2016, 1860, 97–107. [Google Scholar] [CrossRef]
- Santofimia-Castano, P.; Rizzuti, B.; Pey, A.L.; Soubeyran, P.; Vidal, M.; Urrutia, R.; Iovanna, J.L.; Neira, J.L. Intrinsically disordered chromatin protein NUPR1 binds to the C-terminal region of Polycomb RING1B. Proc. Natl. Acad. Sci. USA 2017, 114, E6332–E6341. [Google Scholar] [CrossRef] [Green Version]
- Santofimia-Castano, P.; Xia, Y.; Lan, W.; Zhou, Z.; Huang, C.; Peng, L.; Soubeyran, P.; Velazquez-Campoy, A.; Abian, O.; Rizzuti, B.; et al. Ligand-based design identifies a potent NUPR1 inhibitor exerting anticancer activity via necroptosis. J. Clin. Investig. 2019, 129, 2500–2513. [Google Scholar] [CrossRef]
- Nolan, K.A.; Scott, K.A.; Barnes, J.; Doncaster, J.; Whitehead, R.C.; Stratford, I.J. Pharmacological inhibitors of NAD(P)H quinone oxidoreductase, NQO1: Structure/activity relationships and functional activity in tumour cells. Biochem. Pharmacol. 2010, 80, 977–981. [Google Scholar] [CrossRef]
- Nolan, K.A.; Zhao, H.; Faulder, P.F.; Frenkel, A.D.; Timson, D.J.; Siegel, D.; Ross, D.; Burke, T.R., Jr.; Stratford, I.J.; Bryce, R.A. Coumarin-based inhibitors of human NAD(P)H:quinone oxidoreductase-1. Identification, structure-activity, off-target effects and in vitro human pancreatic cancer toxicity. J. Med. Chem. 2007, 50, 6316–6325. [Google Scholar] [CrossRef]
- Scott, K.A.; Barnes, J.; Whitehead, R.C.; Stratford, I.J.; Nolan, K.A. Inhibitors of NQO1: Identification of compounds more potent than dicoumarol without associated off-target effects. Biochem. Pharmacol. 2011, 81, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Asher, G.; Lotem, J.; Cohen, B.; Sachs, L.; Shaul, Y. Regulation of p53 stability and p53-dependent apoptosis by NADH quinone oxidoreductase 1. Proc. Natl. Acad. Sci. USA 2001, 98, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Lind, C.; Cadenas, E.; Hochstein, P.; Ernster, L. DT-diaphorase: Purification, properties, and function. Methods Enzymol. 1990, 186, 287–301. [Google Scholar] [PubMed]
- Ernster, L.; Danielson, L.; Ljunggren, M. DT diaphorase. I. Purification from the soluble fraction of rat-liver cytoplasm, and properties. Biochim. Biophys. Acta 1962, 58, 171–188. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. Functions of NQO1 in Cellular Protection and CoQ10 Metabolism and its Potential Role as a Redox Sensitive Molecular Switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef]
- Beyer, R.E.; Segura-Aguilar, J.; Di Bernardo, S.; Cavazzoni, M.; Fato, R.; Fiorentini, D.; Galli, M.C.; Setti, M.; Landi, L.; Lenaz, G. The role of DT-diaphorase in the maintenance of the reduced antioxidant form of coenzyme Q in membrane systems. Proc. Natl. Acad. Sci. USA 1996, 93, 2528–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onyenwoke, R.U.; Wiegel, J. Iron (III) reduction: A novel activity of the human NAD(P)H:oxidoreductase. Biochem. Biophys. Res. Commun. 2007, 353, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Haefeli, R.H.; Erb, M.; Gemperli, A.C.; Robay, D.; Courdier Fruh, I.; Anklin, C.; Dallmann, R.; Gueven, N. NQO1-dependent redox cycling of idebenone: Effects on cellular redox potential and energy levels. PLoS ONE 2011, 6, e17963. [Google Scholar] [CrossRef] [Green Version]
- Walton, M.I.; Smith, P.J.; Workman, P. The role of NAD(P)H: Quinone reductase (EC 1.6.99.2, DT-diaphorase) in the reductive bioactivation of the novel indoloquinone antitumor agent EO9. Cancer Commun. 1991, 3, 199–206. [Google Scholar] [CrossRef]
- Brunmark, A.; Cadenas, E.; Lind, C.; Segura-Aguilar, J.; Ernster, L. DT-diaphorase-catalyzed two-electron reduction of quinone epoxides. Free Radic. Biol. Med. 1987, 3, 181–188. [Google Scholar] [CrossRef]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C.; Varnes, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J. Biol. Chem. 2000, 275, 5416–5424. [Google Scholar] [CrossRef] [Green Version]
- Silvers, M.A.; Deja, S.; Singh, N.; Egnatchik, R.A.; Sudderth, J.; Luo, X.; Beg, M.S.; Burgess, S.C.; DeBerardinis, R.J.; Boothman, D.A.; et al. The NQO1 bioactivatable drug, beta-lapachone, alters the redox state of NQO1+ pancreatic cancer cells, causing perturbation in central carbon metabolism. J. Biol. Chem. 2017, 292, 18203–18216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.; Gibson, N.W.; Preusch, P.C.; Ross, D. Metabolism of mitomycin C by DT-diaphorase: Role in mitomycin C-induced DNA damage and cytotoxicity in human colon carcinoma cells. Cancer Res. 1990, 50, 7483–7489. [Google Scholar] [PubMed]
- Siegel, D.; Beall, H.; Senekowitsch, C.; Kasai, M.; Arai, H.; Gibson, N.W.; Ross, D. Bioreductive activation of mitomycin C by DT-diaphorase. Biochemistry 1992, 31, 7879–7885. [Google Scholar] [CrossRef] [PubMed]
- Nemeikaite-Ceniene, A.; Sarlauskas, J.; Jonusiene, V.; Maroziene, A.; Miseviciene, L.; Yantsevich, A.V.; Cenas, N. Kinetics of Flavoenzyme-Catalyzed Reduction of Tirapazamine Derivatives: Implications for Their Prooxidant Cytotoxicity. Int. J. Mol. Sci. 2019, 20, 4602. [Google Scholar] [CrossRef] [Green Version]
- Sarlauskas, J.; Miseviciene, L.; Maroziene, A.; Karvelis, L.; Stankeviciute, J.; Krikstopaitis, K.; Cenas, N.; Yantsevich, A.; Laurynenas, A.; Anusevicius, Z. The study of NADPH-dependent flavoenzyme-catalyzed reduction of benzo [1,2-c]1,2,5-oxadiazole N-oxides (benzofuroxans). Int. J. Mol. Sci. 2014, 15, 23307–23331. [Google Scholar] [CrossRef] [Green Version]
- Miseviciene, L.; Anusevicius, Z.; Sarlauskas, J.; Cenas, N. Reduction of nitroaromatic compounds by NAD(P)H:quinone oxidoreductase (NQO1): The role of electron-accepting potency and structural parameters in the substrate specificity. Acta Biochim. Pol. 2006, 53, 569–576. [Google Scholar] [CrossRef]
- Sarlauskas, J.; Dickancaite, E.; Nemeikaite, A.; Anusevicius, Z.; Nivinskas, H.; Segura-Aguilar, J.; Cenas, N. Nitrobenzimidazoles as substrates for DT-diaphorase and redox cycling compounds: Their enzymatic reactions and cytotoxicity. Arch. Biochem. Biophys. 1997, 346, 219–229. [Google Scholar] [CrossRef]
- Munoz, P.; Cardenas, S.; Huenchuguala, S.; Briceno, A.; Couve, E.; Paris, I.; Segura-Aguilar, J. DT-Diaphorase Prevents Aminochrome-Induced Alpha-Synuclein Oligomer Formation and Neurotoxicity. Toxicol. Sci. 2015, 145, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Graumann, R.; Paris, I.; Martinez-Alvarado, P.; Rumanque, P.; Perez-Pastene, C.; Cardenas, S.P.; Marin, P.; Diaz-Grez, F.; Caviedes, R.; Caviedes, P.; et al. Oxidation of dopamine to aminochrome as a mechanism for neurodegeneration of dopaminergic systems in Parkinson’s disease. Possible neuroprotective role of DT-diaphorase. Pol. J. Pharmacol. 2002, 54, 573–579. [Google Scholar]
- Hosoda, S.; Nakamura, W.; Hayashi, K. Properties and reaction mechanism of DT diaphorase from rat liver. J. Biol. Chem. 1974, 249, 6416–6423. [Google Scholar] [CrossRef]
- Rase, B.; Bartfai, T.; Ernster, L. Purification of DT-diaphorase by affinity chromatography. Occurrence of two subunits and nonlinear Dixon and Scatchard plots of the inhibition by anticoagulants. Arch. Biochem. Biophys. 1976, 172, 380–386. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Asher, G.; Reiss, V.; Shaul, Y.; Sachs, L.; Lotem, J. Inhibition of NAD(P)H:quinone oxidoreductase 1 activity and induction of p53 degradation by the natural phenolic compound curcumin. Proc. Natl. Acad. Sci. USA 2005, 102, 5535–5540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patino-Morales, C.C.; Soto-Reyes, E.; Arechaga-Ocampo, E.; Ortiz-Sanchez, E.; Antonio-Vejar, V.; Pedraza-Chaverri, J.; Garcia-Carranca, A. Curcumin stabilizes p53 by interaction with NAD(P)H:quinone oxidoreductase 1 in tumor-derived cell lines. Redox Biol. 2020, 28, 101320. [Google Scholar] [CrossRef] [PubMed]
- Megarity, C.F.; Timson, D.J. Cancer-associated variants of human NQO1: Impacts on inhibitor binding and cooperativity. Biosci. Rep. 2019, 39, BSR20191874. [Google Scholar] [CrossRef] [Green Version]
- Tie, J.K.; Jin, D.Y.; Straight, D.L.; Stafford, D.W. Functional study of the vitamin K cycle in mammalian cells. Blood 2011, 117, 2967–2974. [Google Scholar] [CrossRef] [Green Version]
- Asher, G.; Lotem, J.; Sachs, L.; Kahana, C.; Shaul, Y. Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQO1. Proc. Natl. Acad. Sci. USA 2002, 99, 13125–13130. [Google Scholar] [CrossRef] [Green Version]
- Anwar, A.; Dehn, D.; Siegel, D.; Kepa, J.K.; Tang, L.J.; Pietenpol, J.A.; Ross, D. Interaction of human NAD(P)H:quinone oxidoreductase 1 (NQO1) with the tumor suppressor protein p53 in cells and cell-free systems. J. Biol. Chem. 2003, 278, 10368–10373. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Pickart, C.M.; Coffino, P. Determinants of proteasome recognition of ornithine decarboxylase, a ubiquitin-independent substrate. EMBO J. 2003, 22, 1488–1496. [Google Scholar] [CrossRef] [Green Version]
- Anwar, A.; Siegel, D.; Kepa, J.K.; Ross, D. Interaction of the molecular chaperone Hsp70 with human NAD(P)H:quinone oxidoreductase 1. J. Biol. Chem. 2002, 277, 14060–14067. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkov, P.; Adamovich, Y.; Elliott, E.; Shaul, Y. E3 ligase STUB1/CHIP regulates NAD(P)H:quinone oxidoreductase 1 (NQO1) accumulation in aged brain, a process impaired in certain Alzheimer disease patients. J. Biol. Chem. 2011, 286, 8839–8845. [Google Scholar] [CrossRef] [Green Version]
- Adler, J.; Reuven, N.; Kahana, C.; Shaul, Y. c-Fos proteasomal degradation is activated by a default mechanism, and its regulation by NAD(P)H:quinone oxidoreductase 1 determines c-Fos serum response kinetics. Mol. Cell. Biol. 2010, 30, 3767–3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yang, L.N.; Cheng, L.; Tu, S.; Guo, S.J.; Le, H.Y.; Xiong, Q.; Mo, R.; Li, C.Y.; Jeong, J.S.; et al. Bcl2-associated athanogene 3 interactome analysis reveals a new role in modulating proteasome activity. Mol. Cell. Proteom. 2013, 12, 2804–2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garate, M.; Wong, R.P.; Campos, E.I.; Wang, Y.; Li, G. NAD(P)H quinone oxidoreductase 1 inhibits the proteasomal degradation of the tumour suppressor p33(ING1b). EMBO Rep. 2008, 9, 576–581. [Google Scholar] [CrossRef] [Green Version]
- Garate, M.; Campos, E.I.; Bush, J.A.; Xiao, H.; Li, G. Phosphorylation of the tumor suppressor p33(ING1b) at Ser-126 influences its protein stability and proliferation of melanoma cells. FASEB J. 2007, 21, 3705–3716. [Google Scholar] [CrossRef] [PubMed]
- Alard, A.; Fabre, B.; Anesia, R.; Marboeuf, C.; Pierre, P.; Susini, C.; Bousquet, C.; Pyronnet, S. NAD(P)H quinone-oxydoreductase 1 protects eukaryotic translation initiation factor 4GI from degradation by the proteasome. Mol. Cell. Biol. 2010, 30, 1097–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, T.; Tanabe-Fujimura, C.; Fujita, Y.; Abe, C.; Nanakida, Y.; Zou, K.; Liu, J.; Liu, S.; Nakajima, T.; Komano, H. NAD(P)H quinone oxidoreductase 1 inhibits the proteasomal degradation of homocysteine-induced endoplasmic reticulum protein. Biochem. Biophys. Res. Commun. 2016, 473, 1276–1280. [Google Scholar] [CrossRef]
- Hori, O.; Ichinoda, F.; Yamaguchi, A.; Tamatani, T.; Taniguchi, M.; Koyama, Y.; Katayama, T.; Tohyama, M.; Stern, D.M.; Ozawa, K.; et al. Role of Herp in the endoplasmic reticulum stress response. Genes Cells 2004, 9, 457–469. [Google Scholar] [CrossRef]
- Adamovich, Y.; Shlomai, A.; Tsvetkov, P.; Umansky, K.B.; Reuven, N.; Estall, J.L.; Spiegelman, B.M.; Shaul, Y. The protein level of PGC-1alpha, a key metabolic regulator, is controlled by NADH-NQO1. Mol. Cell. Biol. 2013, 33, 2603–2613. [Google Scholar] [CrossRef] [Green Version]
- Guryanova, O.A.; Drazba, J.A.; Frolova, E.I.; Chumakov, P.M. Actin cytoskeleton remodeling by the alternatively spliced isoform of PDLIM4/RIL protein. J. Biol. Chem. 2011, 286, 26849–26859. [Google Scholar] [CrossRef] [Green Version]
- Hershkovitz Rokah, O.; Shpilberg, O.; Granot, G. NAD(P)H quinone oxidoreductase protects TAp63gamma from proteasomal degradation and regulates TAp63gamma-dependent growth arrest. PLoS ONE 2010, 5, e11401. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salido, E.; Timson, D.J.; Betancor-Fernández, I.; Palomino-Morales, R.; Anoz-Carbonell, E.; Pacheco-García, J.L.; Medina, M.; Pey, A.L. Targeting HIF-1α Function in Cancer through the Chaperone Action of NQO1: Implications of Genetic Diversity of NQO1. J. Pers. Med. 2022, 12, 747. https://doi.org/10.3390/jpm12050747
Salido E, Timson DJ, Betancor-Fernández I, Palomino-Morales R, Anoz-Carbonell E, Pacheco-García JL, Medina M, Pey AL. Targeting HIF-1α Function in Cancer through the Chaperone Action of NQO1: Implications of Genetic Diversity of NQO1. Journal of Personalized Medicine. 2022; 12(5):747. https://doi.org/10.3390/jpm12050747
Chicago/Turabian StyleSalido, Eduardo, David J. Timson, Isabel Betancor-Fernández, Rogelio Palomino-Morales, Ernesto Anoz-Carbonell, Juan Luis Pacheco-García, Milagros Medina, and Angel L. Pey. 2022. "Targeting HIF-1α Function in Cancer through the Chaperone Action of NQO1: Implications of Genetic Diversity of NQO1" Journal of Personalized Medicine 12, no. 5: 747. https://doi.org/10.3390/jpm12050747