
Targeted RNAseq Improves Clinical Diagnosis of Very Early-Onset Pediatric Immune Dysregulation

 , ,
, , 
Abstract
:1. Introduction
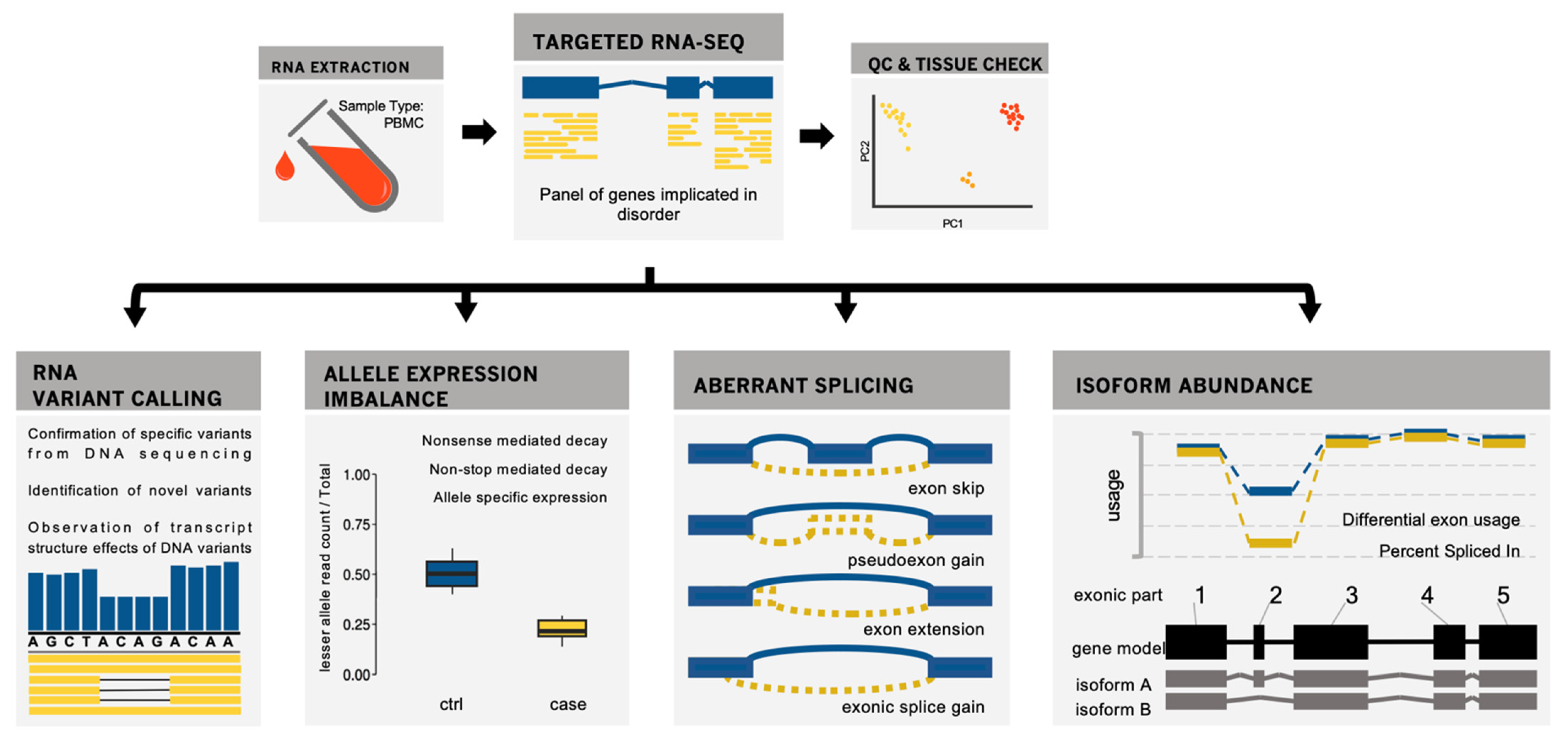
2. Materials and Methods
2.1. Sequencing
2.2. Alignment and Pre-Processing
2.3. Variant Calling
2.4. Allele Expression Imbalance
2.5. Exon Usage Analysis
2.6. Splicing
2.7. Complementary Analysis
3. Results
3.1. Development of a Targeted RNAseq Panel for Immunodeficiency Analysis
3.2. Application to Seven Cases of Primary Immunodeficiency
3.3. Application to Six Cases of Very Early-Onset Inflammatory Bowel Disease
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suwinski, P.; Ong, C.; Ling, M.H.T.; Poh, Y.M.; Khan, A.M.; Ong, H.S. Advancing Personalized Medicine Through the Application of Whole Exome Sequencing and Big Data Analytics. Front. Genet. 2019, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Burgess, D.J. The TOPMed genomic resource for human health. Nat. Rev. Genet. 2021, 22, 200. [Google Scholar] [CrossRef]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Ankala, A.; Ms, C.D.S.; Gualandi, F.; Ferlini, A.; Bean, L.J.H.; Collins, C.; Tanner, A.K.; Hegde, M.R. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann. Neurol. 2015, 77, 206–214. [Google Scholar] [CrossRef]
- Cummings, B.B.; Marshall, J.L.; Tukiainen, T.; Lek, M.; Donkervoort, S.; Foley, A.R.; Bolduc, V.; Waddell, L.B.; Sandaradura, S.A.; O’Grady, G.L.; et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 2017, 9, eaal5209. [Google Scholar] [CrossRef] [Green Version]
- Dimmock, D.; Caylor, S.; Waldman, B.; Benson, W.; Ashburner, C.; Carmichael, J.L.; Carroll, J.; Cham, E.; Chowdhury, S.; Cleary, J.; et al. Project Baby Bear: Rapid precision care incorporating rWGS in 5 California children’s hospitals demonstrates improved clinical outcomes and reduced costs of care. Am. J. Hum. Genet. 2021, 108, 1231–1238. [Google Scholar] [CrossRef]
- Grosse, S.D.; Rasmussen, S.A. Exome sequencing: Value is in the eye of the beholder. Genet. Med. 2020, 22, 280–282. [Google Scholar] [CrossRef]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef]
- Marshall, C.R.; Initiative, O.B.O.T.M.G.; Bick, D.; Belmont, J.W.; Taylor, S.L.; Ashley, E.; Dimmock, D.; Jobanputra, V.; Kearney, H.M.; Kulkarni, S.; et al. The Medical Genome Initiative: Moving whole-genome sequencing for rare disease diagnosis to the clinic. Genome Med. 2020, 12, 1–4. [Google Scholar] [CrossRef]
- Rockowitz, S.; LeCompte, N.; Carmack, M.; Quitadamo, A.; Wang, L.; Park, M.; Knight, D.; Sexton, E.; Smith, L.; Sheidley, B.; et al. Children’s rare disease cohorts: An integrative research and clinical genomics initiative. NPJ Genom. Med. 2020, 5, 29. [Google Scholar] [CrossRef]
- Shin, H.Y.; Jang, H.; Han, J.H.; Park, H.J.; Lee, J.H.; Kim, S.W.; Kim, S.M.; Park, Y.-E.; Kim, D.-S.; Bang, D.; et al. Targeted next-generation sequencing for the genetic diagnosis of dysferlinopathy. Neuromuscul. Disord. 2015, 25, 502–510. [Google Scholar] [CrossRef]
- Wright, C.F.; Fitzgerald, T.W.; Jones, W.D.; Clayton, S.; McRae, J.F.; van Kogelenberg, M.; King, D.A.; Ambridge, K.; Barrett, D.M.; Bayzetinova, T.; et al. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome-wide research data. Lancet 2015, 385, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.F.; FitzPatrick, D.R.; Firth, H.V. Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 2018, 19, 253–268. [Google Scholar] [CrossRef]
- Marcon, A.R.; Bieber, M.; Caulfield, T. Representing a “revolution”: How the popular press has portrayed personalized medicine. Genet. Med. 2018, 20, 950–956. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.R.; Lorenzo, D.; Salerno, J.; Yeh, V.M.; Mitrani, V.B.; Kripalani, S. Current applications of precision medicine: A bibliometric analysis. Pers. Med. 2019, 16, 351–359. [Google Scholar] [CrossRef]
- Momozawa, Y.; Mizukami, K. Unique roles of rare variants in the genetics of complex diseases in humans. J. Hum. Genet. 2021, 66, 11–23. [Google Scholar] [CrossRef]
- Bousfiha, A.; Jeddane, L.; Picard, C.; Ailal, F.; Bobby Gaspar, H.; Al-Herz, W.; Chatila, T.; Crow, Y.J.; Cunningham-Rundles, C.; Etzioni, A.; et al. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J. Clin. Immunol. 2018, 38, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Modell, V.; Orange, J.S.; Quinn, J.; Modell, F. Global report on primary immunodeficiencies: 2018 update from the Jeffrey Modell Centers Network on disease classification, regional trends, treatment modalities, and physician reported outcomes. Immunol. Res. 2018, 66, 367–380. [Google Scholar] [CrossRef]
- Picard, C.; Bobby Gaspar, H.; Al-Herz, W.; Bousfiha, A.; Casanova, J.L.; Chatila, T.; Crow, Y.J.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J. Clin. Immunol. 2018, 38, 96–128. [Google Scholar] [CrossRef] [Green Version]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020, 40, 24–64. [Google Scholar] [CrossRef] [Green Version]
- Barmada, A.; Ramaswamy, A.; Lucas, C.L. Maximizing insights from monogenic immune disorders. Curr. Opin. Immunol. 2021, 73, 50–57. [Google Scholar] [CrossRef]
- Gruber, C.; Bogunovic, D. Incomplete penetrance in primary immunodeficiency: A skeleton in the closet. Qual. Life Res. 2020, 139, 745–757. [Google Scholar] [CrossRef]
- Flinn, A.M.; Gennery, A.R. Primary immune regulatory disorders: Undiagnosed needles in the haystack? Orphanet J. Rare Dis. 2022, 17, 99. [Google Scholar] [CrossRef]
- De Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.-G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Kugathasan, S.; Denson, L.A.; Walters, T.D.; Kim, M.-O.; Marigorta, U.M.; Schirmer, M.; Kajari Mondal, K.; Liu, C.; Griffiths, A.; Noe, J.D.; et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: A multicentre inception cohort study. Lancet 2017, 389, 1710–1718. [Google Scholar] [CrossRef] [Green Version]
- Kerur, B.; I Benchimol, E.; Fiedler, K.; Stahl, M.; Hyams, J.; Stephens, M.; Lu, Y.; Pfefferkorn, M.; Alkhouri, R.; Strople, J.; et al. Natural History of Very Early Onset Inflammatory Bowel Disease in North America: A Retrospective Cohort Study. Inflamm. Bowel Dis. 2021, 27, 295–302. [Google Scholar] [CrossRef]
- Zheng, H.B.; De La Morena, M.T.; Suskind, D.L. The Growing Need to Understand Very Early Onset Inflammatory Bowel Disease (VEO-IBD). Front. Immunol. 2021, 12, 1858. [Google Scholar] [CrossRef]
- Ouahed, J.; Spencer, E.; Kotlarz, D.; Shouval, D.S.; Kowalik, M.; Peng, K.; Field, M.; Grushkin-Lerner, L.; Pai, S.-Y.; Bousvaros, A.; et al. Very Early Onset Inflammatory Bowel Disease: A Clinical Approach With a Focus on the Role of Genetics and Underlying Immune Deficiencies. Inflamm. Bowel Dis. 2020, 26, 820–842. [Google Scholar] [CrossRef]
- Uhlig, H.H.; Schwerd, T.; Koletzko, S.; Shah, N.; Kammermeier, J.; Elkadri, A.; Ouahed, J.; Wilson, D.C.; Travis, S.P.; Turner, D.; et al. The Diagnostic Approach to Monogenic Very Early Onset Inflammatory Bowel Disease. Gastroenterology 2014, 147, 990–1007.e3. [Google Scholar] [CrossRef] [Green Version]
- Hyams, J.S.; Thomas, S.D.; Gotman, N.; Haberman, Y.; Karns, R.; Schirmer, M.; Mo, A.; Mack, D.R.; Boyle, B.; Griffiths, A.M.; et al. Clinical and biological predictors of response to standardised paediatric colitis therapy (PROTECT): A multicentre inception cohort study. Lancet 2019, 393, 1708–1720. [Google Scholar] [CrossRef]
- Kelsen, J.R.; Dawany, N.; Moran, C.J.; Petersen, B.-S.; Sarmady, M.; Sasson, A.; Pauly-Hubbard, H.; Martinez, A.; Maurer, K.; Soong, J.; et al. Exome sequencing analysis reveals variants in primary immunodeficiency genes in patients with very early onset inflammatory bowel disease. Gastroenterology 2015, 149, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Moran, C.J.; Walters, T.D.; Guo, C.-H.; Kugathasan, S.; Klein, C.; Turner, D.; Wolters, V.M.; Bandsma, R.H.; Mouzaki, M.; Zachos, M.; et al. IL-10R Polymorphisms Are Associated with Very-early-onset Ulcerative Colitis. Inflamm. Bowel Dis. 2013, 19, 115–123. [Google Scholar] [CrossRef]
- Digby-Bell, J.L.; Atreya, R.; Monteleone, G.; Powell, N. Interrogating host immunity to predict treatment response in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 9–20. [Google Scholar] [CrossRef]
- Condino-Neto, A.; Espinosa-Rosales, F.J. Changing the Lives of People With Primary Immunodeficiencies (PI) With Early Testing and Diagnosis. Front. Immunol. 2018, 9, 1439. [Google Scholar] [CrossRef]
- Bhuvanagiri, M.; Schlitter, A.M.; Hentze, M.W.; Kulozik, A.E. NMD: RNA biology meets human genetic medicine. Biochem. J. 2010, 430, 365–377. [Google Scholar] [CrossRef] [Green Version]
- Chabot, B.; Shkreta, L. Defective control of pre–messenger RNA splicing in human disease. J. Cell Biol. 2016, 212, 13–27. [Google Scholar] [CrossRef]
- Curry, P.D.K.; Broda, K.L.; Carroll, C.J. The Role of RNA-Sequencing as a New Genetic Diagnosis Tool. Curr. Genet. Med. Rep. 2021, 9, 13–21. [Google Scholar] [CrossRef]
- Gonorazky, H.D.; Naumenko, S.; Ramani, A.K.; Nelakuditi, V.; Mashouri, P.; Wang, P.; Kao, D.; Ohri, K.; Viththiyapaskaran, S.; Tarnopolsky, M.A.; et al. Expanding the boundaries of RNA sequencing as a diagnostic tool for rare Mendelian disease. Am. J. Hum. Genet. 2019, 104, 466–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, J.; Baralle, D. Splicing in the Diagnosis of Rare Disease: Advances and Challenges. Front. Genet. 2021, 12, 689892. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.; Aloraini, T.; Al Subaie, L.; Alissa, A.; Al Qudsi, A.; Alahmad, A.; Al Mutairi, F.; Alswaid, A.; Alothaim, A.; Eyaid, W.; et al. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet. Med. 2018, 20, 1328–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costain, G.; Jobling, R.; Walker, S.; Reuter, M.S.; Snell, M.; Bowdin, S.; Cohn, R.D.; Dupuis, L.; Hewson, S.; Mercimek-Andrews, S.; et al. Periodic reanalysis of whole-genome sequencing data enhances the diagnostic advantage over standard clinical genetic testing. Eur. J. Hum. Genet. 2018, 26, 740–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Meng, L.; Normand, E.A.; Xia, F.; Song, X.; Ghazi, A.; Rosenfeld, J.; Magoulas, P.L.; Braxton, A.; Ward, P.; et al. Reanalysis of Clinical Exome Sequencing Data. N. Engl. J. Med. 2019, 380, 2478–2480. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Guturu, H.; Bernstein, J.A.; Bejerano, G. Systematic reanalysis of clinical exome data yields additional diagnoses: Implications for providers. Genet. Med. 2017, 19, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.F.; McRae, J.F.; Clayton, S.; Gallone, G.; Aitken, S.; FitzGerald, T.W.; Jones, P.; Prigmore, E.; Rajan, D.; Lord, J.; et al. Making new genetic diagnoses with old data: Iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet. Med. 2018, 20, 1216–1223. [Google Scholar] [CrossRef] [Green Version]
- Al-Murshedi, F.; Meftah, D.; Scott, P. Underdiagnoses resulting from variant misinterpretation: Time for systematic reanalysis of whole exome data? Eur. J. Med. Genet. 2019, 62, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Basel-Salmon, L.; Orenstein, N.; Markus-Bustani, K.; Ruhrman-Shahar, N.; Kilim, Y.; Magal, N.; Hubshman, M.W.; Bazak, L. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet. Med. 2019, 21, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Ardigó, D. Addressing challenges in the diagnosis and treatment of rare genetic diseases. Nat. Rev. Drug Discov. 2018, 17, 151–152. [Google Scholar] [CrossRef] [PubMed]
- Frésard, L.; Smail, C.; Ferraro, N.M.; Teran, N.A.; Li, X.; Smith, K.S.; Bonner, D.; Kernohan, K.D.; Marwaha, S.; Zappala, Z.; et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat. Med. 2019, 25, 911–919. [Google Scholar] [CrossRef]
- Zhang, P.; Itan, Y. Biological Network Approaches and Applications in Rare Disease Studies. Genes 2019, 10, 797. [Google Scholar] [CrossRef] [Green Version]
- Castel, S.E.; Mohammadi, P.; Chung, W.K.; Shen, Y.; Lappalainen, T. Rare variant phasing and haplotypic expression from RNA sequencing with phASER. Nat. Commun. 2016, 7, 12817. [Google Scholar] [CrossRef] [Green Version]
- Chakravorty, S.; Berger, K.; Rufibach, L.; Gloster, L.; Emmons, S.; Shenoy, S.; Hegde, M.; Dinasarapu, A.R.; Gibson, G. Combinatorial clinically driven blood biomarker functional genomics significantly enhances genotype-phenotype resolution and diagnostics in neuromuscular disease. medRxiv 2021. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Hartley, S.W.; Mullikin, J.C. QoRTs: A comprehensive toolset for quality control and data processing of RNA-Seq experiments. BMC Bioinform. 2015, 16, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouard, J.-S.; Schenkel, F.; Marete, A.; Bissonnette, N. The GATK joint genotyping workflow is appropriate for calling variants in RNA-seq experiments. J. Anim. Sci. Biotechnol. 2019, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Gelfman, S.; Wang, Q.; McSweeney, K.M.; Ren, Z.; La Carpia, F.; Halvorsen, M.; Schoch, K.; Ratzon, F.; Heinzen, E.L.; Boland, M.J.; et al. Annotating pathogenic non-coding variants in genic regions. Nat. Commun. 2017, 8, 236. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, S.; Miao, K.; Benson, C.C.; Heinig, M.; Cook, S.A.; Hubner, N. Alternative Splicing Signatures in RNA-seq Data: Percent Spliced in (PSI). Curr. Protoc. Hum. Genet. 2015, 87, 11.16.1–11.16.14. [Google Scholar] [CrossRef]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Rowlands, C.F.; Taylor, A.; Rice, G.; Whiffin, N.; Hall, H.N.; Newman, W.G.; Black, G.C.; O’Keefe, R.T.; Hubbard, S.; Douglas, A.G.; et al. MRSD: A quantitative approach for assessing suitability of RNA-seq in the investigation of mis-splicing in Mendelian disease. Am. J. Hum. Genet. 2022, 109, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Waugh, J.; Perry, C.M. Anakinra: A review of its use in the management of rheumatoid arthritis. BioDrugs 2005, 19, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, S.; Fondanèche, M.-C.; Lambert, N.C.; Pasquier, B.; Mateo, V.; Soulas-Sprauel, P.; Galicier, L.; Le Deist, F.; Rieux-Laucat, F.; Revy, P.; et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 2006, 444, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Moreland, L.; Bate, G.; Kirkpatrick, P. Fresh from the pipeline: Abatacept. Nat. Rev. Drug Discov. 2006, 5, 185–186. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenton, S.B.; Partovi, N.; Ensom, M.H. Sirolimus: The evidence for clinical pharmacokinetic monitoring. Clin. Pharmacokinet. 2005, 44, 769–786. [Google Scholar] [CrossRef]
- Dorjbal, B.; Stinson, J.R.; Ma, C.A.; Weinreich, M.A.; Miraghazadeh, B.; Hartberger, J.M.; Frey-Jakobs, S.; Weidinger, S.; Moebus, L.; Andre Franke, A.; et al. Hypomorphic caspase activation and recruitment domain 11 (CARD11) mutations associated with diverse immunologic phenotypes with or without atopic disease. J. Allergy Clin. Immunol. 2019, 143, 1482–1495. [Google Scholar] [CrossRef] [Green Version]
- Salah, S.; Hegazy, R.; Ammar, R.; Sheba, H.; AbdelRahman, L. MEFV gene mutations and cardiac phenotype in children with familial Mediterranean fever: A cohort study. Pediatr. Rheumatol. 2014, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Federici, S.; Calcagno, G.; Finetti, M.; Gallizzi, R.; Meini, A.; Vitale, A.; Caroli, F.; Cattalini, M.; Caorsi, R.; Zulian, F.; et al. Clinical impact of MEFV mutations in children with periodic fever in a prevalent western European Caucasian population. Ann. Rheum. Dis. 2012, 71, 1961–1965. [Google Scholar] [CrossRef]
- Moradian, M.M.; Sarkisian, T.; Amaryan, G.; Hayrapetyan, H.; Yeghiazaryan, A.; Davidian, N.; Avanesian, N. Patient management and the association of less common familial Mediterranean fever symptoms with other disorders. Genet. Med. 2014, 16, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lei, F.; Wang, X.; Deng, K.; Ji, Y.; Zhang, Y.; Li, H.; Zhang, X.; Lu, Z.; Zhang, P. NULP1 Alleviates Cardiac Hypertrophy by Suppressing NFAT3 Transcriptional Activity. J. Am. Heart Assoc. 2020, 9, e016419. [Google Scholar] [CrossRef] [PubMed]
- Schnappauf, O.; Heale, L.; Dissanayake, D.; Tsai, W.L.; Gadina, M.; Leto, T.L.; Kastner, D.L.; Malech, H.L.; Kuhns, D.B.; Aksentijevich, I.; et al. Homozygous variant p. Arg90His in NCF1 is associated with early-onset Interferonopathy: A case report. Pediatr. Rheumatol. 2021, 19, 54. [Google Scholar] [CrossRef] [PubMed]
- Wrona, D.; Siler, U.; Reichenbach, J. Novel Diagnostic Tool for p47phox-Deficient Chronic Granulomatous Disease Patient and Carrier Detection. Mol. Ther. Methods Clin. Dev. 2019, 13, 274–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacout, C.; Haddad, E.; Sabri, S.; Svinarchouk, F.; Garçon, L.; Capron, C.; Foudi, A.; Mzali, R.; Snapper, S.B.; Louache, F.; et al. A defect in hematopoietic stem cell migration explains the nonrandom X-chromosome inactivation in carriers of Wiskott-Aldrich syndrome. Blood 2003, 102, 1282–1289. [Google Scholar] [CrossRef]
- Lutskiy, M.I.; Sasahara, Y.; Kenney, D.M.; Rosen, F.S.; Remold-O’Donnell, E. Wiskott-Aldrich syndrome in a female. Blood J. Am. Soc. Hematol. 2002, 100, 2763–2768. [Google Scholar] [CrossRef]
- Denson, L.A.; Jurickova, I.; Karns, R.; Shaw, K.A.; Cutler, D.J.; Okou, D.T.; Dodd, A.; Quinn, K.; Mondal, K.; Aronow, B.J.; et al. Clinical and Genomic Correlates of Neutrophil Reactive Oxygen Species Production in Pediatric Patients With Crohn’s Disease. Gastroenterology 2018, 154, 2097–2110. [Google Scholar] [CrossRef]
- Ruan, J.; Schlüter, D.; Naumann, M.; Waisman, A.; Wang, X. Ubiquitin-modifying enzymes as regulators of colitis. Trends Mol. Med. 2022, 28, 304–318. [Google Scholar] [CrossRef]
- Hu, B.; Zhang, D.; Zhao, K.; Wang, Y.; Pei, L.; Fu, Q.; Ma, X. Spotlight on USP4: Structure, function, and regulation. Front. Cell Dev. Biol. 2021, 148, 595159. [Google Scholar] [CrossRef]
- De Valles-Ibáñez, G.; Esteve-Sole, A.; Piquer, M.; González-Navarro, E.A.; Hernandez-Rodriguez, J.; Laayouni, H.; González-Roca, E.; Plaza-Martin, A.M.; Deyà-Martínez, Á.; Martín-Nalda, A.; et al. Evaluating the genetics of common variable immunodeficiency: Monogenetic model and beyond. Front. Immunol. 2018, 9, 636. [Google Scholar] [CrossRef]
- Ostrowski, J.; Paziewska, A.; Lazowska-Przeorek, I.; Ambrożkiewicz, F.; Goryca, K.; Kulecka, M.; Rawa, T.; Karczmarski, J.; Dąbrowska, M.; Żeber-Lubecka, N.; et al. Genetic architecture differences between pediatric and adult-onset inflammatory bowel diseases in the Polish population. Sci. Rep. 2016, 6, 39831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuthbert, A.P.; Fisher, S.A.; Mirza, M.M.; King, K.; Hampe, J.; Croucher, P.J.; Mascheretti, S.; Sanderson, J.; Forbes, A.; Mansfield, J.; et al. The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology 2002, 122, 867–874. [Google Scholar] [CrossRef]
- Lesage, S.; Zouali, H.; Cézard, J.-P.; Colombel, J.-F.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.; Gassull, M.; Binder, V.; et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am. J. Hum. Genet. 2002, 70, 845–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.H.; Abraham, C. Inflammatory Bowel Disease Genetics: Nod2. Annu. Rev. Med. 2007, 58, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, J.E.; Warner, N.; Staples, J.; Crowley, E.; Gosalia, N.; Murchie, R.; Van Hout, C.; Fiedler, K.; Welch, G.; King, A.K.; et al. Mutation spectrum of NOD2 reveals recessive inheritance as a main driver of Early Onset Crohn’s Disease. Sci. Rep. 2021, 11, 5595. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, H.; Vitali, M.; Lougaris, V.; Giliani, S.; Parvaneh, N.; Parvaneh, L.; Mirminachi, B.; Cheraghi, T.; Khazaei, H.; Mahdaviani, S.A.; et al. Cohort of Iranian Patients with Congenital Agammaglobulinemia: Mutation Analysis and Novel Gene Defects. Expert Rev. Clin. Immunol. 2016, 12, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, R.; Abolhassani, H.; Kiaee, F.; Habibi, S.; Azizi, G.; Tavakol, M.; Chavoshzadeh, Z.; Mahdaviani, S.A.; Momen, T.; Gharagozlou, M.; et al. Comparison of Common Monogenic Defects in a Large Predominantly Antibody Deficiency Cohort. J. Allergy Clin. Immunol. Pract. 2019, 7, 864–878.e9. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Zhou, M.; Uckun, F.M. Crystal structure of Bruton’s tyrosine kinase domain suggests a novel pathway for activation and provides insights into the molecular basis of X-linked agammaglobulinemia. J. Biol. Chem. 2001, 276, 41435–41443. [Google Scholar] [CrossRef] [Green Version]
- Väliaho, J.; Faisal, I.; Ortutay, C.; Smith, C.I.E.; Vihinen, M. Characterization of All Possible Single-Nucleotide Change Caused Amino Acid Substitutions in the Kinase Domain of Bruton Tyrosine Kinase. Hum. Mutat. 2015, 36, 638–647. [Google Scholar] [CrossRef]
- Uhlig, H.H. Monogenic diseases associated with intestinal inflammation: Implications for the understanding of inflammatory bowel disease. Gut 2013, 62, 1795–1805. [Google Scholar] [CrossRef]
- Yin, Q.; Lin, S.-C.; Lo, Y.-C.; Damo, S.M.; Wu, H. Tumor Necrosis Factor Receptor-Associated Factors in Immune Receptor Signal Transduction. In Handbook of Cell Signaling; Elsevier: Amsterdam, The Netherlands, 2010; pp. 339–345. [Google Scholar]
- De Diego, R.P.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Plancoulaine, S.; Picard, C.; Herman, M.; Cardon, A.; Durandy, A.; Bustamante, J.; et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 2010, 33, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Cleary, A.M.; Ye, Z.S.; Hong, D.I.; Lederman, S.; Baltimore, D. Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science 1995, 267, 1494–1498. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Pietras, E.M.; He, J.Q.; Kang, J.R.; Liu, S.-Y.; Oganesyan, G.; Shahangian, A.; Zarnegar, B.; Shiba, T.L.; Wang, Y.; et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006, 25, 3257–3263. [Google Scholar] [CrossRef]
- Brodeur, S.R.; Cheng, G.; Baltimore, D.; Thorley-Lawson, D.A. Localization of the major NF-κB-activating site and the sole TRAF3 binding site of LMP-1 defines two distinct signaling motifs. J. Biol. Chem. 1997, 272, 19777–19784. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Pan, W.; Shi, P.; Gao, H.; Zhao, F.; Song, X.; Liu, Y.; Zhao, L.; Li, X.; Shi, Y.; et al. Modulation of experimental autoimmune encephalomyelitis through TRAF3-mediated suppression of interleukin 17 receptor signaling. J. Exp. Med. 2010, 207, 2647–2662. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Choi, J.-K.; Li, M.; Kaye, K.; Kieff, E.; Jung, J.U. Role of cellular tumor necrosis factor receptor-associated factors in NF-κB activation and lymphocyte transformation by herpesvirus saimiri STP. J. Virol. 1999, 73, 3913–3919. [Google Scholar] [CrossRef] [Green Version]
- Dyle, M.C.; Kolakada, D.; Cortazar, M.A.; Jagannathan, S. How to get away with nonsense: Mechanisms and consequences of escape from nonsense-mediated RNA decay. Wiley Interdiscip. Rev. RNA 2020, 11, e1560. [Google Scholar] [CrossRef]
- Haddow, J.B.; Musbahi, O.; Macdonald, T.T.; Knowles, C.H. Comparison of cytokine and phosphoprotein profiles in idiopathic and Crohn’s disease-related perianal fistula. World J. Gastrointest. Pathophysiol. 2019, 10, 42–53. [Google Scholar] [CrossRef]
- Hucthagowder, V.; Meyer, R.; Mullins, C.; Nagarajan, R.; DiPersio, J.F.; Vij, R.; Tomasson, M.H.; Kulkarni, S. Resequencing analysis of the candidate tyrosine kinase and RAS pathway gene families in multiple myeloma. Cancer Genet. 2012, 205, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.; Francioli, L. The Genome Aggregation Database (Gnomad); MacArthur Lab: Sydney, Australia, 2017. [Google Scholar]
- Rothlin, C.V.; Leighton, J.A.; Ghosh, S. Tyro3, Axl, and Mertk Receptor Signaling in Inflammatory Bowel Disease and Colitis-associated Cancer. Inflamm. Bowel Dis. 2014, 20, 1472–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, V.; Banday, A.Z.; Jindal, A.K.; Das, J.; Rawat, A. Recent advances in elucidating the genetics of common variable immunodeficiency. Genes Dis. 2020, 7, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.L.; Chandra, A.; Nejentsev, S.; Condliffe, A.M.; Okkenhaug, K. PI3Kδ and primary immunodeficiencies. Nat. Rev. Immunol. 2016, 16, 702–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swan, D.J.; Aschenbrenner, D.; Lamb, C.A.; Chakraborty, K.; Clark, J.; Pandey, S.; Engelhardt, K.R.; Chen, R.; Cavounidis, A.; Ding, Y.; et al. Immunodeficiency, autoimmune thrombocytopenia and enterocolitis caused by autosomal recessive deficiency of PIK3CD-encoded phosphoinositide 3-kinase δ. Haematologica 2019, 104, e483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Rotival, M.; Patin, E.; Michel, F.; Pellegrini, S. Two common disease-associated TYK2 variants impact exon splicing and TYK2 dosage. PLoS ONE 2020, 15, e0225289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Góth, L.; Rass, P.; Madarasi, I. A novel catalase mutation detected by polymerase chain reaction-single strand conformation polymorphism, nucleotide sequencing, and Western blot analyses is responsible for the type C of Hungarian acatalasemia. Electrophoresis 2001, 22, 49–51. [Google Scholar] [CrossRef]
- Gifford, C.A.; Ranade, S.S.; Samarakoon, R.; Salunga, H.T.; de Soysa, T.Y.; Huang, Y.; Zhou, P.; Elfenbein, A.; Wyman, S.K.; Bui, Y.K.; et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 2019, 364, 865–870. [Google Scholar] [CrossRef]
- Rahme, E.; Joseph, L. Estimating the prevalence of a rare disease: Adjusted maximum likelihood. J. R. Stat. Soc. Ser. D Stat. 1998, 47, 149–158. [Google Scholar] [CrossRef]
- Auvin, S.; Irwin, J.; Abi-Aad, P.; Battersby, A. The Problem of Rarity: Estimation of Prevalence in Rare Disease. Value Health 2018, 21, 501–507. [Google Scholar] [CrossRef] [Green Version]
- Goth, L.; Nagy, T. Inherited catalase deficiency: Is it benign or a factor in various age related disorders? Mutat. Res. Rev. Mutat. Res. 2013, 753, 147–154. [Google Scholar] [CrossRef]
- Hamilton, H.B.; Neel, J.V.; Kobara, T.Y.; Ozaki, K. The frequency in Japan of carriers of the rare “recessive” gene causing acatalasemia. J. Clin. Investig. 1961, 40, 2199–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertes, C.; Scheller, I.F.; Yépez, V.A.; Çelik, M.H.; Liang, Y.; Kremer, L.S.; Gusic, M.; Prokisch, H.; Gagneur, J. Detection of aberrant splicing events in RNA-seq data using FRASER. Nat. Commun. 2021, 12, 529. [Google Scholar] [CrossRef] [PubMed]
- Schlieben, L.D.; Prokisch, H.; Yépez, V.A. How Machine Learning and Statistical Models Advance Molecular Diagnostics of Rare Disorders Via Analysis of RNA Sequencing Data. Front. Mol. Biosci. 2021, 8, 647277. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Gene | HGVS | Predicted AA Change | Variant Type | Zygosity | Splice Effect in RNA | rs no. | SIFT | PolyPhen2 | CADD Phred | Interpro Domain | GnomAD MAF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P25 | MEFV | c.2040G>C | p.M680I | missense | HET | - | rs28940580 | Tolerated | Benign | 0.002 | SPRY | 0.0001 |
| CARD11 | c.224G>A | p.R75Q | missense | HET | - | rs1064795280 | Damaging | Prob.Dam. | 33 | CARD | - | |
| TCF25 | unknown | HET | intron retention | - | - | - | - | - | - | |||
| P49 | CTLA4 | c.442C>T | p.Q148X | nonsense | HET | - | - | - | - | 38 | Ig-like | - |
| P55 | NCF2 | c.812A>G | p.K271R | missense | HET | - | - | Tolerated | Prob.Dam. | 24.4 | SH3 | - |
| NCF1 | c.269G>A | p.R90H | missense | HET | - | rs201802880 | Damaging | Benign | 25 | Phox homolog | 0.001 | |
| WAS | c.689AGA{2} | p.K232del | nonframeshift del | HET | - | rs782409127 | - | - | - | - | 9.67 × 10−5 | |
| P69 | XIAP | gross del | HEMI | x4-5 skip | - | - | - | - | - | |||
| MERTK | c.844G>A | p.A282T | missense/ splice | HET | leaky exon skip | rs7588635 | Damaging | Benign | 24.2 | Ig-like | 0.0108 (AFR: 0.14) | |
| P89 | NCF1 | c.247G>A | p.G83R | missense | HET | - | rs139225348 | Tolerated | Poss.Dam. | 20.2 | Phox homolog | 0.0089 |
| CHB535 | TRAF3 | c.1275C>G | p.Y425X | nonsense | HET | - | - | - | - | 35 | MATH/TRAF | - |
| CHB749 | BTK | c.1955T>C | p.L652P | missense | HEMI | - | rs128622212 | Tolerated | Poss.Dam. | 20.6 | Protein kinase | - |
| CHB786 | NOD2 | c.2722G>C | p.G908R | missense | HET | - | rs2066845 | Damaging | Prob.Dam. | 31 | Leu-rich repeat | 0.0113 |
| ERBIN | c.3704A>C | p.Y1235S | missense | HET | - | rs201900105 | Tolerated | Benign | 19.43 | - | 1.39 × 10−5 | |
| RBCK1 | c.992C>T | p.S331L | missense | HET | - | rs781592121 | Damaging | Benign | 26.3 | Zinc finger | 9.37 × 10−5 | |
| TYK2 | c.2456G>A | p.S819N | missense | HET | - | rs763006605 | Tolerated | Benign | 7.356 | Protein kinase | 5.03 × 10−5 | |
| CHB953 | PIK3CD | c.1595delG | p.W532X | nonsense | HET | - | - | - | - | - | - | - |
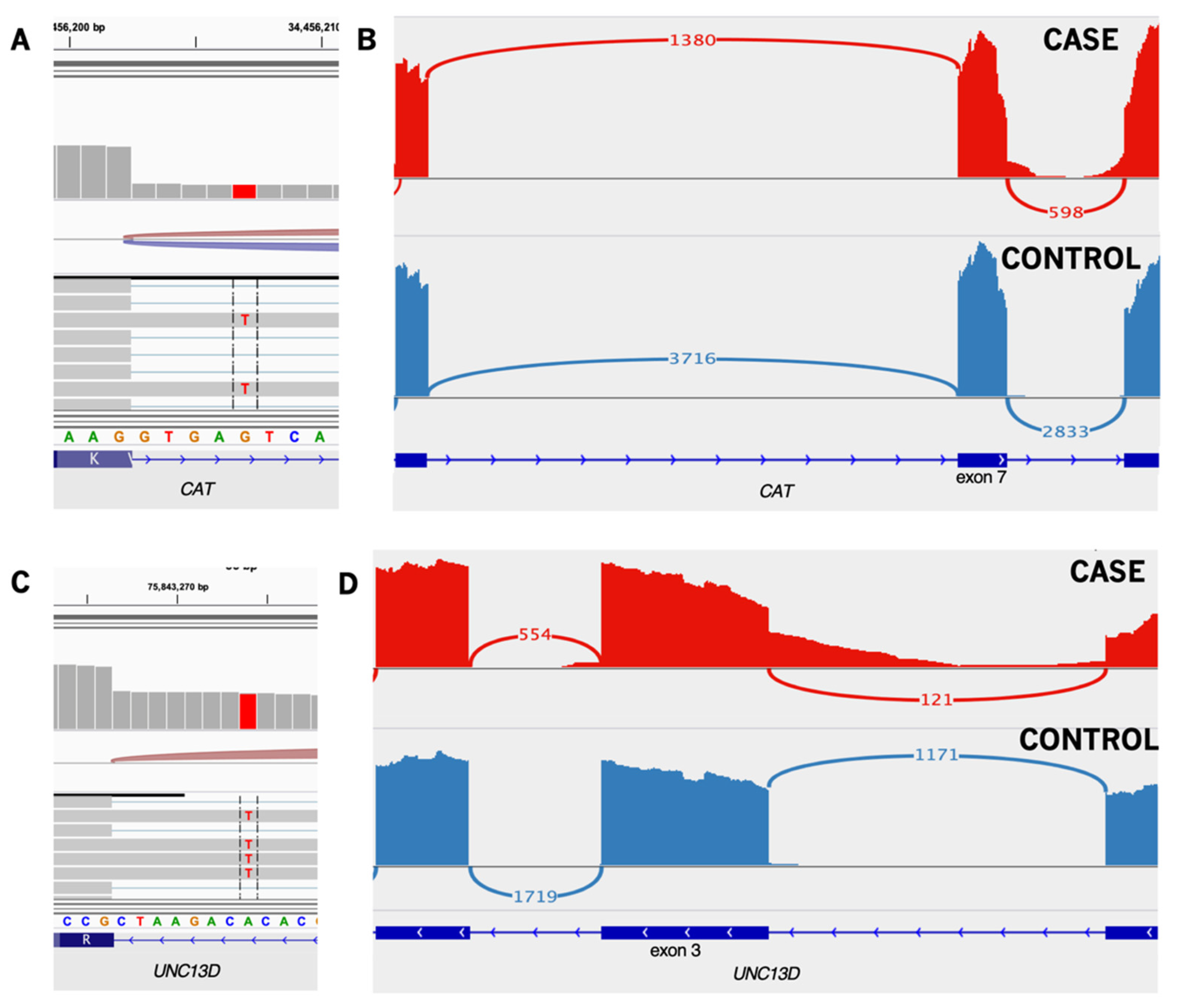
| CAT | c.903+5G>T | - | splice | HET | intron retention | rs147912187 | - | - | - | - | 0.0029 (SAS: 0.01) | |
| UNC13D | c.154-8T>A | - | splice | HET | intron retention | rs369433391 | - | - | - | - | 0.0027 (SAS: 0.01) | |
| TYK2 | c.1001_1011+4del | p.V333_E337del | nonframeshift del | HET | - | - | - | - | - | FERM | 2.63 × 10−5 | |
| CHB974 | NOD2 | c.3017dupC | p.A1006fs | frameshift ins | HET | - | rs2066847 | - | - | - | - | 0.0151 |
| NOD2 | c.2722G>C | p.G908R | missense | HET | - | rs2066845 | Damaging | Prob.Dam. | 31 | Leu-rich repeat | 0.0113 | |
| CHB1025 | MERTK | c.844G>A | p.A282T | missense/splice | HET | leaky exon skip | rs7588635 | Damaging | Benign | 24.2 | Ig-like | 0.0108 (AFR: 0.14) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berger, K.; Arafat, D.; Chandrakasan, S.; Snapper, S.B.; Gibson, G. Targeted RNAseq Improves Clinical Diagnosis of Very Early-Onset Pediatric Immune Dysregulation. J. Pers. Med. 2022, 12, 919. https://doi.org/10.3390/jpm12060919
Berger K, Arafat D, Chandrakasan S, Snapper SB, Gibson G. Targeted RNAseq Improves Clinical Diagnosis of Very Early-Onset Pediatric Immune Dysregulation. Journal of Personalized Medicine. 2022; 12(6):919. https://doi.org/10.3390/jpm12060919
Chicago/Turabian StyleBerger, Kiera, Dalia Arafat, Shanmuganathan Chandrakasan, Scott B. Snapper, and Greg Gibson. 2022. "Targeted RNAseq Improves Clinical Diagnosis of Very Early-Onset Pediatric Immune Dysregulation" Journal of Personalized Medicine 12, no. 6: 919. https://doi.org/10.3390/jpm12060919