
Development of a Machine Learning Model to Predict Non-Durable Response to Anti-TNF Therapy in Crohn’s Disease Using Transcriptome Imputed from Genotypes

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Genotyping
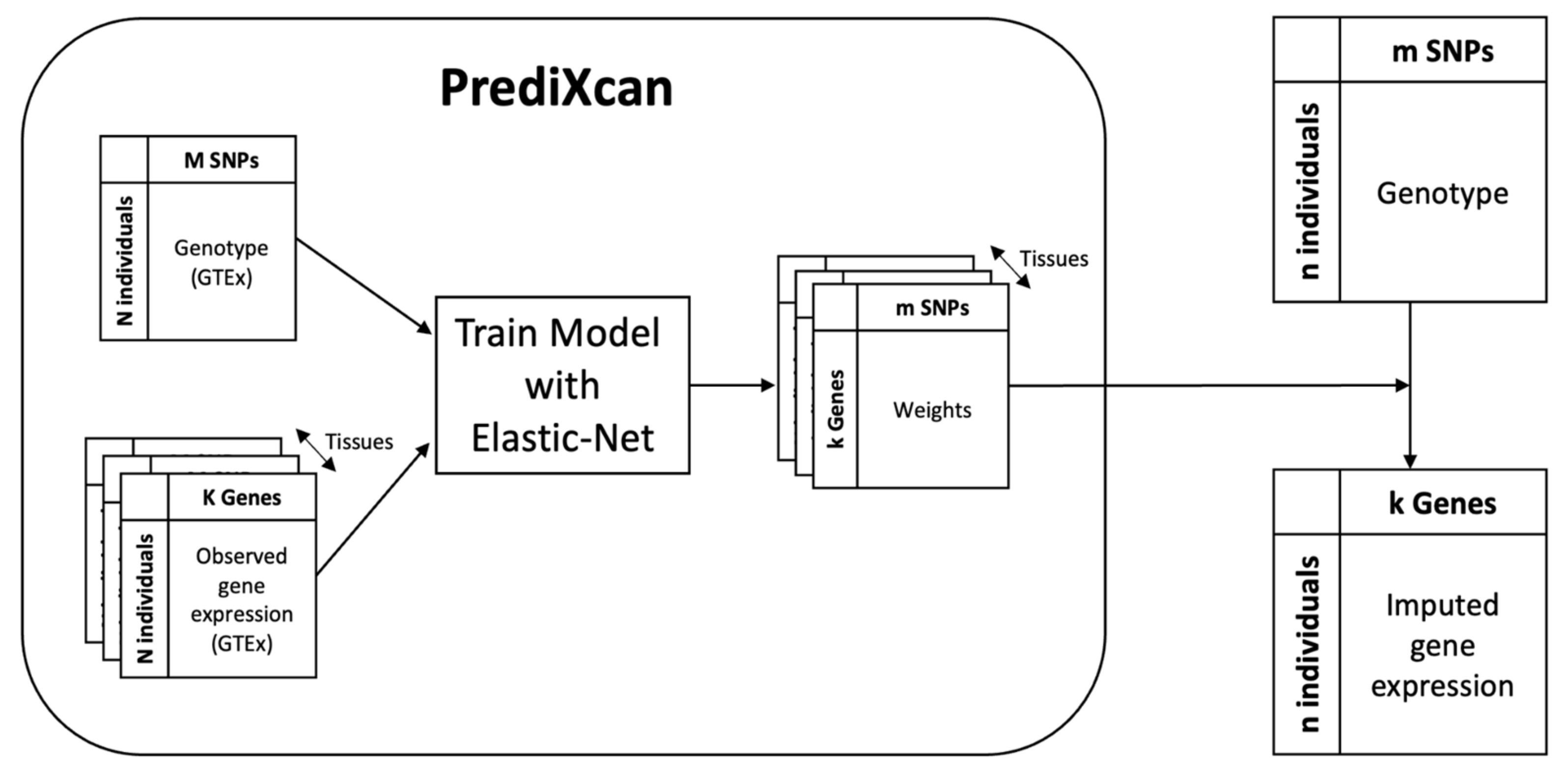
2.3. Imputing Gene Expression from Genotype
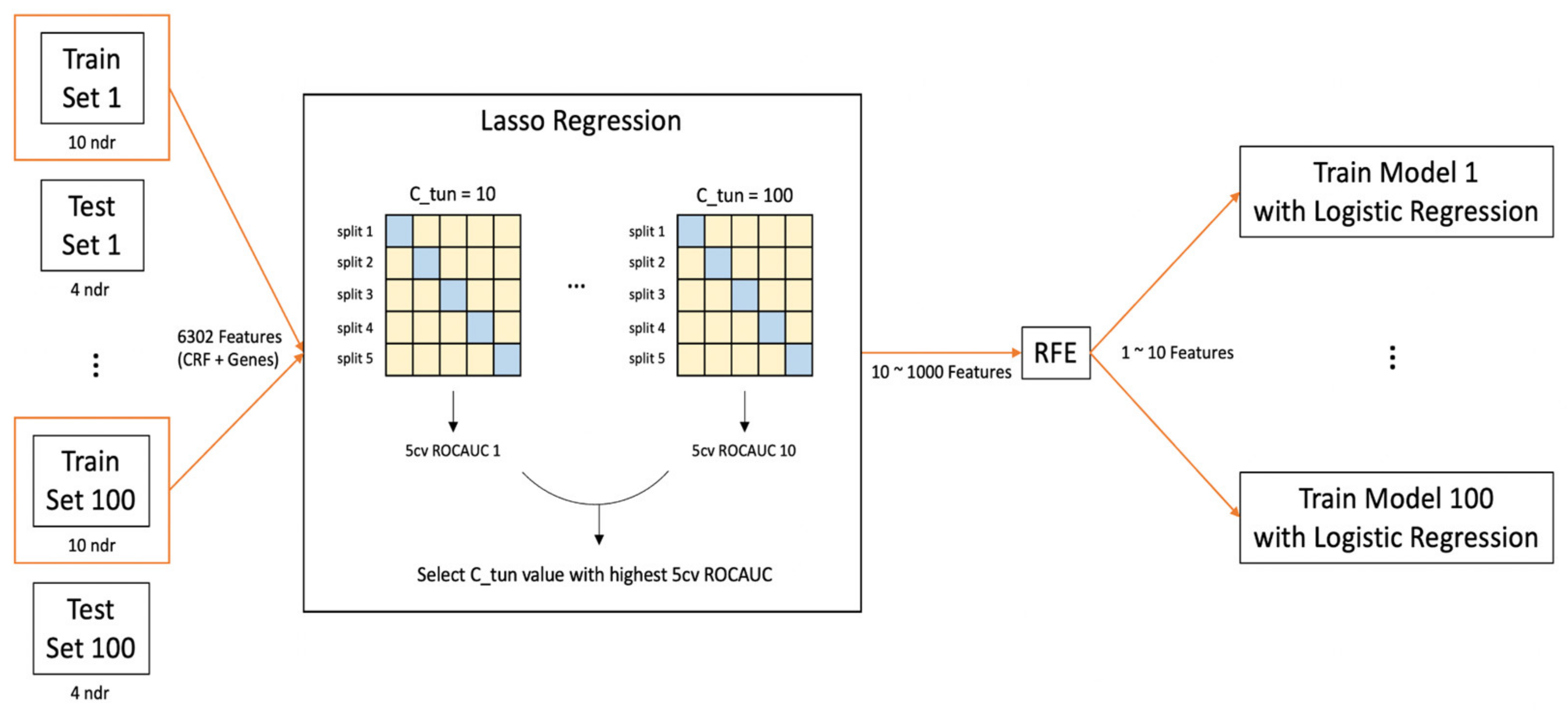
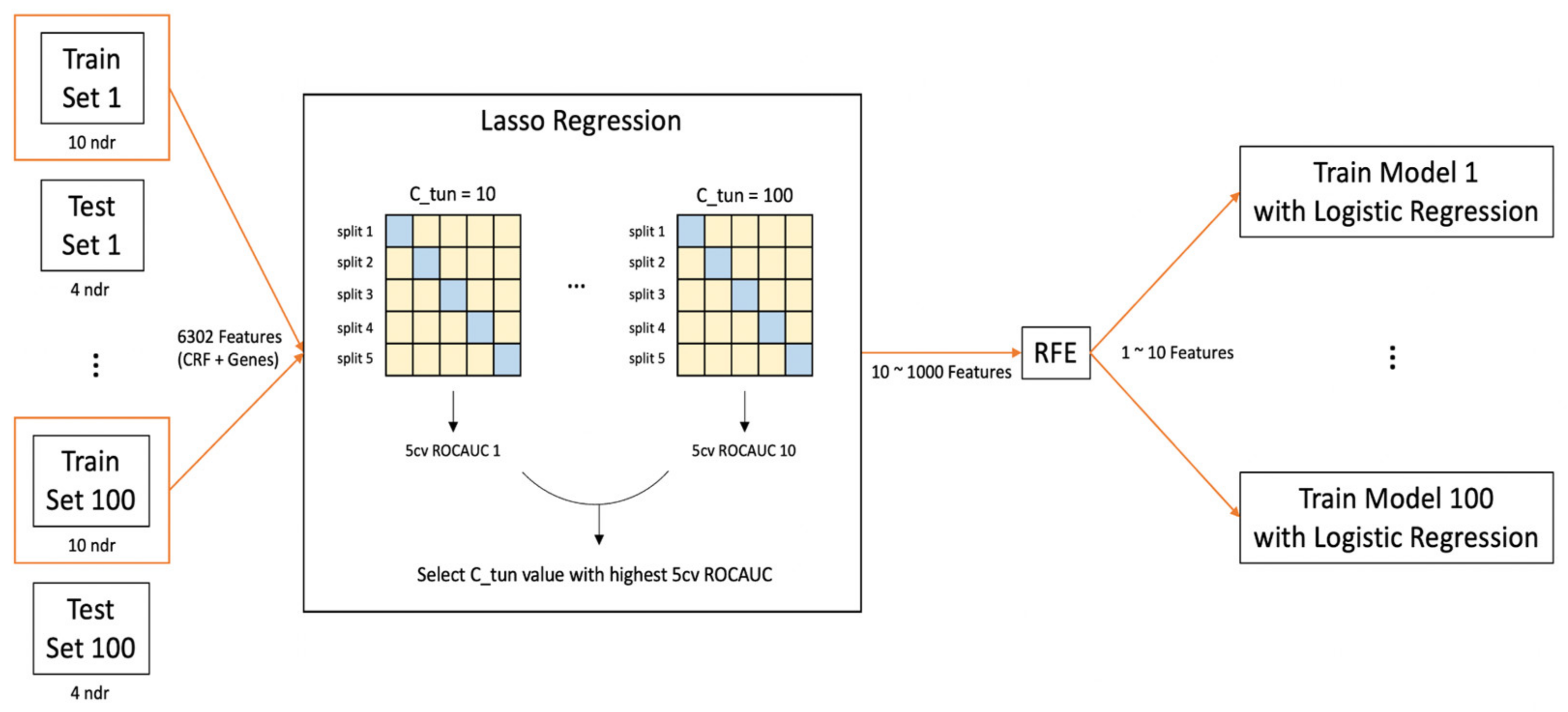
2.4. Development of Predictive Machine Learning Models
2.4.1. Preprocessing
2.4.2. Model Training and Feature Selection
3. Results
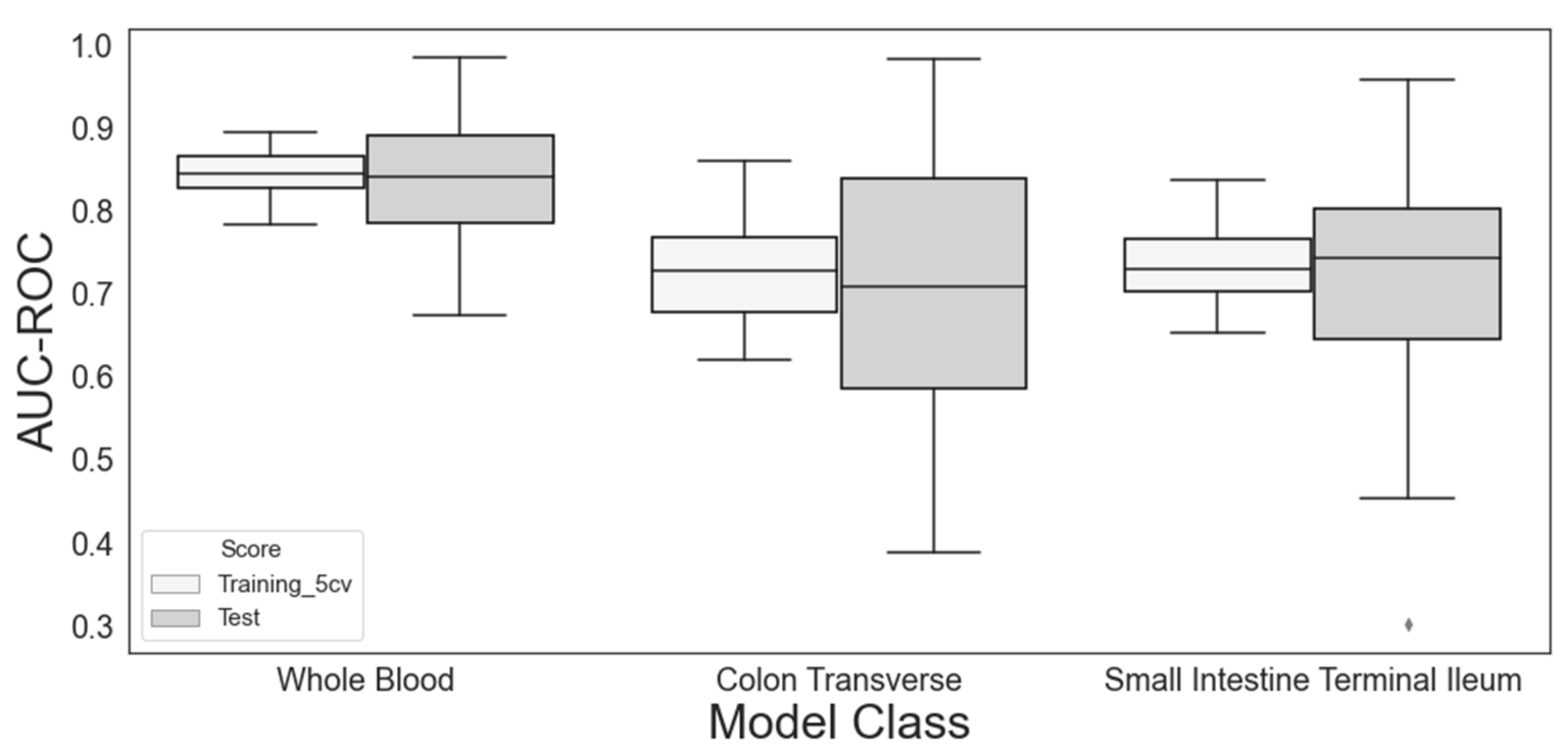
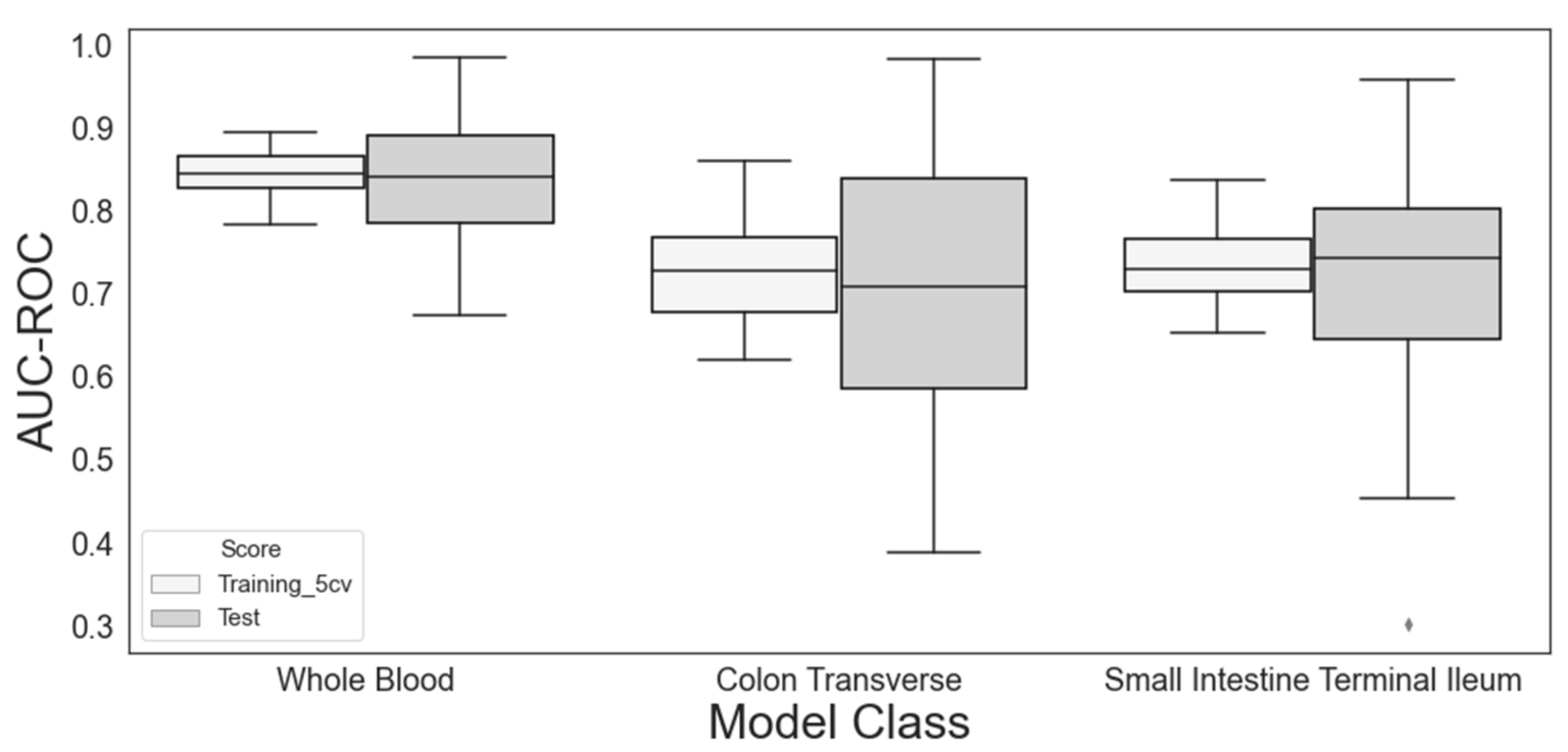
3.1. DR/NDR Prediction Models Based on Different Sampling Tissues
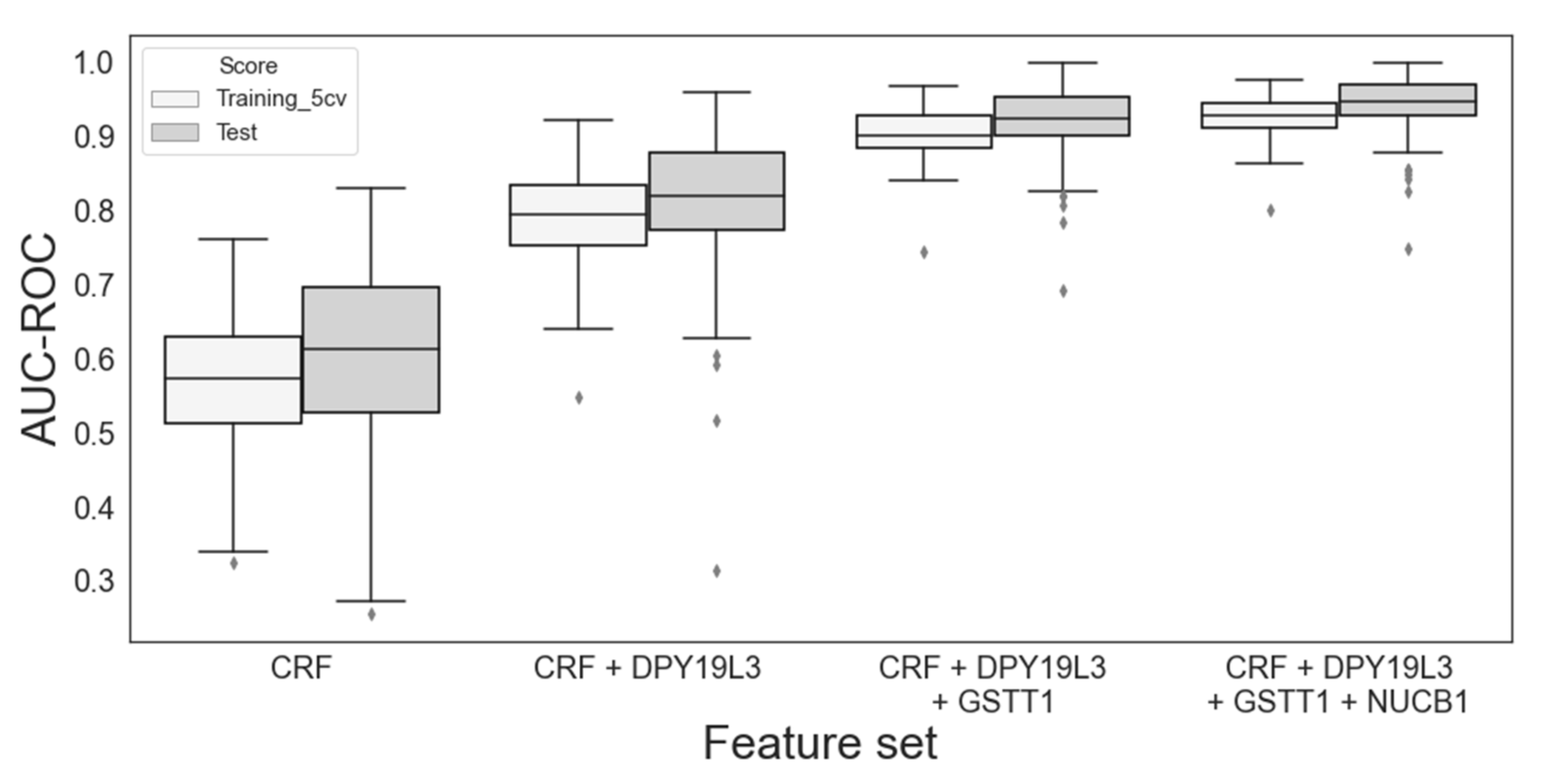
3.2. Selection of Top Two and Three Features Using Whole-Blood Model
3.3. Contribution of Clinical Features
3.4. Genetic Bases of Selected Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cosnes, J.; Gower–Rousseau, C.; Seksik, P.; Cortot, A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 2011, 140, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- D’Haens, G.R.; Panaccione, R.; Higgins, P.D.; Vermeire, S.; Gassull, M.; Chowers, Y.; Hanauer, S.B.; Herfarth, H.; Hommes, D.W.; Kamm, M.; et al. The London Position Statement of the World Congress of Gastroenterology on Biological Therapy for IBD with the European Crohn’s and Colitis Organization: When to start, when to stop, which drug to choose, and how to predict response? Am. J. Gastroenterol. 2011, 106, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Allez, M.; Karmiris, K.; Louis, E.; Van Assche, G.; Ben-Horin, S.; Klein, A.; Van der Woude, J.; Baert, F.; Eliakim, R.; Katsanos, K.; et al. Report of the ECCO pathogenesis workshop on anti-TNF therapy failures in inflammatory bowel diseases: Definitions, frequency and pharmacological aspects. J. Crohn’s Colitis 2010, 4, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Chowers, Y.; Sturm, A.; Sans, M.; Papadakis, K.; Gazouli, M.; Harbord, M.; Jahnel, J.; Mantzaris, G.J.; Meier, J.; Mottet, C.; et al. Report of the ECCO workshop on anti-TNF therapy failures in inflammatory bowel diseases: Biological roles and effects of TNF and TNF antagonists. J. Crohn’s Colitis 2010, 4, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Gisbert, J.P.; Panés, J. Loss of response and requirement of infliximab dose intensification in Crohn’s disease: A review. Am. J. Gastroenterol. 2009, 104, 760–767. [Google Scholar] [CrossRef]
- Hlavaty, T.; Pierik, M.; Henckaerts, L.; Ferrante, M.; Joossens, S.; Van Schuerbeek, N.; Noman, M.; Rutgeerts, P.; Vermeire, S. Polymorphisms in apoptosis genes predict response to infliximab therapy in luminal and fistulizing Crohn’s disease. Aliment. Pharmacol. Ther. 2005, 22, 613–626. [Google Scholar] [CrossRef]
- Jürgens, M.; Laubender, R.P.; Hartl, F.; Weidinger, M.; Seiderer, J.; Wagner, J.; Wetzke, M.; Beigel, F.; Pfennig, S.; Stallhofer, J.; et al. Disease activity, ANCA, and IL23R genotype status determine early response to infliximab in patients with ulcerative colitis. Am. J. Gastroenterol. 2010, 105, 1811–1819. [Google Scholar] [CrossRef]
- Mascheretti, S.; Hampe, J.; Kühbacher, T.; Herfarth, H.; Krawczak, M.; Fölsch, U.R.; Schreiber, S. Pharmacogenetic investigation of the TNF/TNF-receptor system in patients with chronic active Crohn’s disease treated with infliximab. Pharm. J. 2002, 2, 127–136. [Google Scholar] [CrossRef]
- Siegel, C.A.; Melmed, G.Y. Predicting response to Anti-TNF Agents for the treatment of crohn’s disease. Ther. Adv. Gastroenterol. 2009, 2, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.D.; Yang, H.; Landers, C.J.; Rotter, J.I.; Targan, S.R.; Plevy, S.E.; Barry, M.J. ANCA pattern and LTA haplotype relationship to clinical responses to anti-TNF antibody treatment in Crohn’s disease. Gastroenterology 2001, 120, 1347–1355. [Google Scholar] [CrossRef]
- Vermeire, S.; Louis, E.; Rutgeerts, P.; De Vos, M.; Van Gossum, A.; Belaiche, J.; Pescatore, P.; Fiasse, R.; Pelckmans, P.; Vlietinck, R.; et al. NOD2/CARD15 does not influence response to infliximab in Crohn’s disease. Gastroenterology 2002, 123, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Verstockt, B.; Verstockt, S.; Dehairs, J.; Ballet, V.; Blevi, H.; Wollants, W.J.; Breynaert, C.; Van Assche, G.; Vermeire, S.; Ferrante, M. Low TREM1 expression in whole blood predicts anti-TNF response in inflammatory bowel disease. EBioMedicine 2019, 40, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Verstockt, B.; Verstockt, S.; Blevi, H.; Cleynen, I.; de Bruyn, M.; Van Assche, G.; Vermeire, S.; Ferrante, M. TREM-1, the ideal predictive biomarker for endoscopic healing in anti-TNF-treated Crohn’s disease patients? Gut 2019, 68, 1531–1533. [Google Scholar] [CrossRef]
- Gaujoux, R.; Starosvetsky, E.; Maimon, N.; Vallania, F.; Bar-Yoseph, H.; Pressman, S.; Weisshof, R.; Goren, I.; Rabinowitz, K.; Waterman, M.; et al. Cell-centred meta-analysis reveals baseline predictors of anti-TNFα non-response in biopsy and blood of patients with IBD. Gut 2019, 68, 604–614. [Google Scholar] [CrossRef]
- Urcelay, E.; Mendoza, J.L.; Martínez, A.; Fernández, L.; Taxonera, C.; Díaz-Rubio, M.; de la Concha, E.G. IBD5 polymorphisms in inflammatory bowel disease: Association with response to infliximab. World J. Gastroenterol. 2005, 11, 1187–1192. [Google Scholar] [CrossRef]
- Louis, E.; El Ghoul, Z.; Vermeire, S.; Dall’Ozzo, S.; Rutgeerts, P.; Paintaud, G.; Belaiche, J.; De Vos, M.; Van Gossum, A.; Colombel, J.F.; et al. Association between polymorphism in IgG Fc receptor IIIa coding gene and biological response to infliximab in Crohn’s disease. Aliment. Pharmacol. Ther. 2004, 19, 511–519. [Google Scholar] [CrossRef]
- Koder, S.; Repnik, K.; Ferkolj, I.; Pernat, C.; Skok, P.; Weersma, R.K.; Potočnik, U. Genetic polymorphism in ATG16L1 gene influences the response to adalimumab in Crohn’s disease patients. Pharmacogenomics 2015, 16, 191–204. [Google Scholar] [CrossRef]
- Park, S.K.; Kim, S.; Lee, G.Y.; Kim, S.Y.; Kim, W.; Lee, C.W.; Park, J.L.; Choi, C.H.; Kang, S.B.; Kim, T.O.; et al. Development of a Machine Learning Model to Distinguish between Ulcerative Colitis and Crohn’s Disease Using RNA Sequencing Data. Diagnostics 2021, 11, 2365. [Google Scholar] [CrossRef]
- Moon, S.; Kim, Y.J.; Han, S.; Hwang, M.Y.; Shin, D.M.; Park, M.Y.; Lu, Y.; Yoon, K.; Jang, H.M.; Kim, Y.K.; et al. The Korea Biobank Array: Design and Identification of Coding Variants Associated with Blood Biochemical Traits. Sci. Rep. 2019, 9, 1382. [Google Scholar] [CrossRef] [Green Version]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Liu, W.; Baek, J.; Moon, J.W.; Ye, B.D.; Lee, H.S.; Park, S.H.; Yang, S.K.; Han, B.; Liu, J.; et al. Expression Quantitative Trait Loci (eQTL) Mapping in Korean Patients with Crohn’s Disease and Identification of Potential Causal Genes Through Integration With Disease Associations. Front. Genet. 2020, 11, 486. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Gamazon, E.R.; Wheeler, H.E.; Shah, K.P.; Mozaffari, S.V.; Aquino-Michaels, K.; Carroll, R.J.; Eyler, A.E.; Denny, J.C.; Nicolae, D.L.; Cox, N.J.; et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhaylova, A.V.; Thornton, T.A. Accuracy of Gene Expression Prediction From Genotype Data With PrediXcan Varies Across and Within Continental Populations. Front. Genet. 2019, 10, 261. [Google Scholar] [CrossRef] [Green Version]
- Niwa, Y.; Suzuki, T.; Dohmae, N.; Simizu, S. Identification of DPY19L3 as the C-mannosyltransferase of R-spondin1 in human cells. Mol. Biol. Cell 2016, 27, 744–756. [Google Scholar] [CrossRef]
- Haseeb, M.; Pirzada, R.H.; Ain, Q.U.; Choi, S. Wnt Signaling in the Regulation of Immune Cell and Cancer Therapeutics. Cells 2019, 8, 1380. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Ahn, J.B.; Kim, D.H.; Kim, S.; Ma, H.W.; Che, X.; Seo, D.H.; Kim, T.I.; Kim, W.H.; Cheon, J.H.; et al. Glutathione S-transferase theta 1 protects against colitis through goblet cell differentiation via interleukin-22. FASEB J. 2020, 34, 3289–3304. [Google Scholar] [CrossRef] [Green Version]
- Aviello, G.; Knaus, U.G. ROS in gastrointestinal inflammation: Rescue or Sabotage? Br. J. Pharmacol. 2017, 174, 1704–1718. [Google Scholar] [CrossRef] [Green Version]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Rebhan, M.; Chalifa-Caspi, V.; Prilusky, J.; Lancet, D. GeneCards: Integrating information about genes, proteins and diseases. Trends Genet. 1997, 13, 163. [Google Scholar] [CrossRef]
- Li, Z.W.; Sun, B.; Gong, T.; Guo, S.; Zhang, J.; Wang, J.; Sugawara, A.; Jiang, M.; Yan, J.; Gurary, A.; et al. GNAI1 and GNAI3 Reduce Colitis-Associated Tumorigenesis in Mice by Blocking IL6 Signaling and Down-regulating Expression of GNAI2. Gastroenterology 2019, 156, 2297–2312. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Durable Response (n = 14) | Durable Response (n = 220) | p-Value | |
|---|---|---|---|
| Age at diagnosis, year (SD) | 26.3 (9.2) | 28.2 (9.1) | 0.45 |
| Gender, male (%) | 8 (57.1%) | 162 (73.6%) | 0.21 |
| History of smoking, n (%) | 5 (35.7%) | 47 (21.4%) | 0.20 |
| Family history of IBD, n (%) | 0 (0%) | 7 (3.2%) | 1.0 |
| Disease duration, year (SD) | 9.1 (5.5) | 7.5 (3.8) | 0.16 |
| Disease location, n (%) | 0.07 | ||
| Ileal | 7 (50%) | 52 (23.6%) | |
| Colonic | 2 (14.3%) | 31 (14.1%) | |
| Ileocolonic | 5 (35.7%) | 137 (62.3%) | |
| Upper GI involvement, n (%) | 0 (0%) | 11 (5.0%) | 0.39 |
| Disease behavior, n (%) | 0.35 | ||
| Inflammatory | 9 (64.3%) | 163 (74.1%) | |
| Stricturing | 1 (7.1%) | 25 (11.4%) | |
| Penetrating | 4 (28.6%) | 32 (14.5%) | |
| Perianal disease, n (%) | 6 (42.9%) | 85 (38.6%) | 0.75 |
| Combination immunosuppressants, n (%) | 10 (71.4%) | 206 (93.6%) | <0.001 |
| Intestinal resection, n (%) | 6 (42.9%) | 61 (27.7%) | 0.22 |
| Tissue Expression Model | Selected Feature | Selection Frequency | AUC-ROC (SD) | |
|---|---|---|---|---|
| Training 5-CV Set | Test Set | |||
| Whole blood | DPY19L3 | 79/100 | 0.845 (0.027) | 0.839 (0.070) |
| Colon transverse | TXNDC16 | 40/100 | 0.728 (0.060) | 0.711 (0.150) |
| Small intestine terminal ileum | ENSG00000270127 | 14/100 | 0.738 (0.050) | 0.720 (0.120) |
| No. of Features | Selected Feature Set | Selection Frequency | AUC-ROC (SD) | |
|---|---|---|---|---|
| Training 5CV Set | Test Set | |||
| 1 | DPY19L3 | 79 | 0.845 (0.027) | 0.839 (0.070) |
| 2 | DPY19L3, GSTT1 | 32 | 0.918 (0.023) | 0.919 (0.040) |
| 3 | DPY19L3, GSTT1, NUCB1 | 9 | 0.935 (0.024) | 0.935 (0.040) |
| Gene Name | Chr | p-Value | β Value |
|---|---|---|---|
| DPY19L3 | 19 | 0.000965 | 2.703 |
| GSTT1 | 22 | 0.00343 | 1.735 |
| NUCB1 | 19 | 0.00684 | −2.142 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.K.; Kim, Y.B.; Kim, S.; Lee, C.W.; Choi, C.H.; Kang, S.-B.; Kim, T.O.; Bang, K.B.; Chun, J.; Cha, J.M.; et al. Development of a Machine Learning Model to Predict Non-Durable Response to Anti-TNF Therapy in Crohn’s Disease Using Transcriptome Imputed from Genotypes. J. Pers. Med. 2022, 12, 947. https://doi.org/10.3390/jpm12060947
Park SK, Kim YB, Kim S, Lee CW, Choi CH, Kang S-B, Kim TO, Bang KB, Chun J, Cha JM, et al. Development of a Machine Learning Model to Predict Non-Durable Response to Anti-TNF Therapy in Crohn’s Disease Using Transcriptome Imputed from Genotypes. Journal of Personalized Medicine. 2022; 12(6):947. https://doi.org/10.3390/jpm12060947
Chicago/Turabian StylePark, Soo Kyung, Yea Bean Kim, Sangsoo Kim, Chil Woo Lee, Chang Hwan Choi, Sang-Bum Kang, Tae Oh Kim, Ki Bae Bang, Jaeyoung Chun, Jae Myung Cha, and et al. 2022. "Development of a Machine Learning Model to Predict Non-Durable Response to Anti-TNF Therapy in Crohn’s Disease Using Transcriptome Imputed from Genotypes" Journal of Personalized Medicine 12, no. 6: 947. https://doi.org/10.3390/jpm12060947
APA StylePark, S. K., Kim, Y. B., Kim, S., Lee, C. W., Choi, C. H., Kang, S. -B., Kim, T. O., Bang, K. B., Chun, J., Cha, J. M., Im, J. P., Kim, M. S., Ahn, K. S., Kim, S. -Y., & Park, D. I. (2022). Development of a Machine Learning Model to Predict Non-Durable Response to Anti-TNF Therapy in Crohn’s Disease Using Transcriptome Imputed from Genotypes. Journal of Personalized Medicine, 12(6), 947. https://doi.org/10.3390/jpm12060947