
Skin Cancer Research Goes Digital: Looking for Biomarkers within the Droplets




Abstract
:1. Introduction
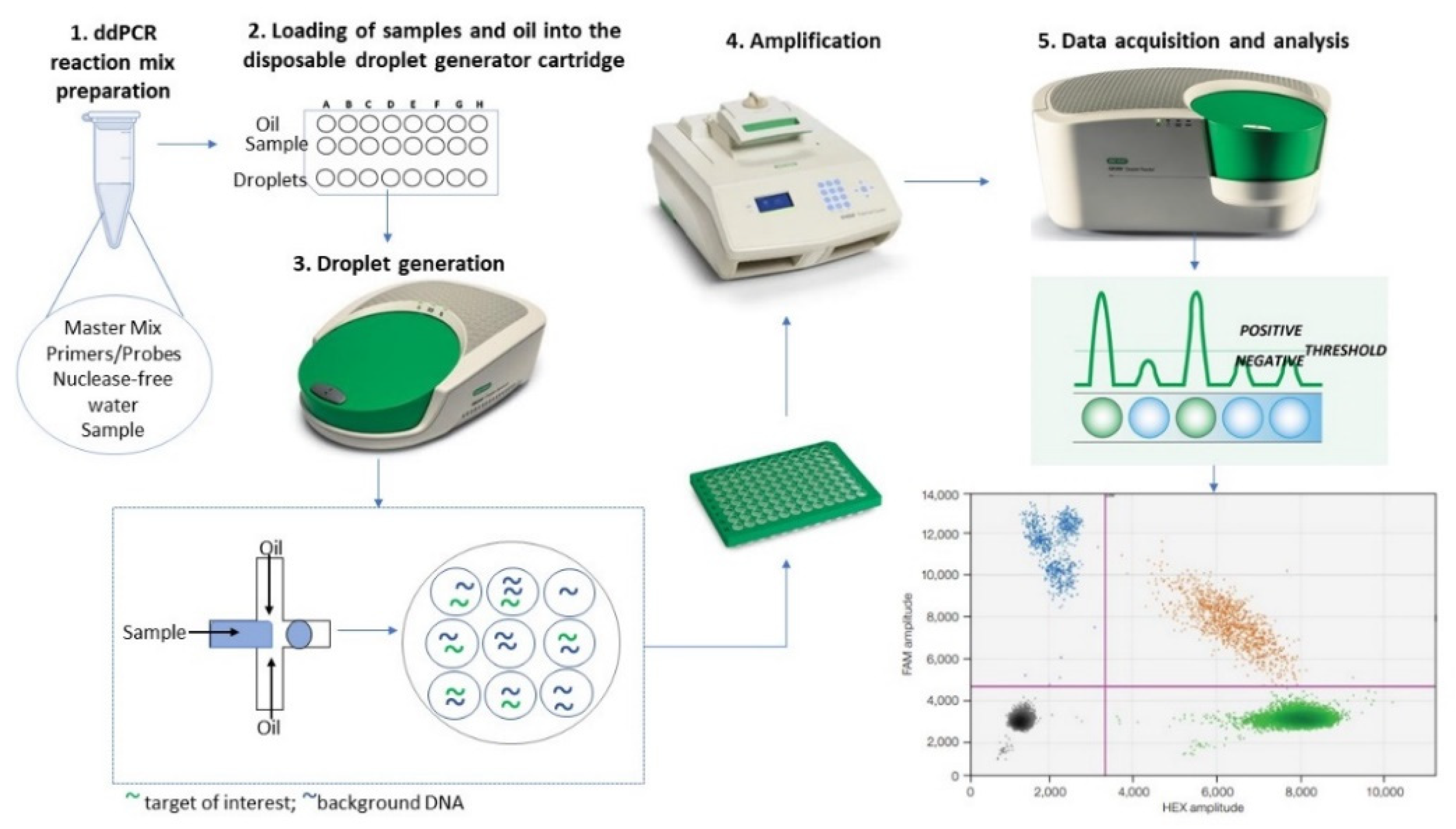
2. The ddPCR Method: A Reliable Omics Technology in Oncology
3. The ddPCR Method for Primary Prevention Strategies and Personalized Skin Cancer Screening
4. The ddPCR Nethod Assisting the Prognosis of Skin Cancer
5. DdPCR-Based Liquid Biopsies for Skin Cancer Monitoring and Post-Treatment Follow-Up
5.1. CtDNA Analysis
5.2. Circulating miRNAs Analysis
5.3. CTCs Analysis
5.4. EVs Analysis
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moon, H.; White, A.C.; Borowsky, A.D. New insights into the functions of Cox-2 in skin and esophageal malignancies. Exp. Mol. Med. 2020, 52, 538–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feller, L.; Khammissa, R.A.G.; Kramer, B.; Altini, M.; Lemmer, J. Basal cell carcinoma, squamous cell carcinoma and melanoma of the head and face. Head Face Med. 2016, 12, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.M.; Lefevre, J.G.; Baxter, G.; Hamilton, N.A. Non-melanoma skin cancer segmentation for histopathology dataset. Data Br. 2021, 39, 107587. [Google Scholar] [CrossRef]
- Fijałkowska, M.; Koziej, M.; Antoszewski, B. Detailed head localization and incidence of skin cancers. Sci. Rep. 2021, 11, 12391. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.Y.; du Preez, D.J.; Millar, D.A.; Norval, M. The Epidemiology of Skin Cancer and Public Health Strategies for Its Prevention in Southern Africa. Int. J. Environ. Res. Public Health 2020, 17, 1017. [Google Scholar] [CrossRef] [Green Version]
- Román-Colón, D.; Rabell-Bernal, A.; Rodriguez-Franco, G.; Santiago-Rivera, L. S3661 A Blurry Surprise: A Case of Metastatic Melanoma. Off. J. Am. Coll. Gastroenterol. ACG 2021, 116, S1495. [Google Scholar] [CrossRef]
- Yadav, P.; Mangal, G.; Bhaumik, U.; Agarwal, S.; Thakor, P.; Lakhanpal, V. Leptomeningeal metastasis from melanoma emulating chronic subdural hematoma: A case report. Egypt. J. Neurosurg. 2021, 36, 35. [Google Scholar] [CrossRef]
- Shetty, A.; Janda, M.; Fry, K.; Brown, S.; Yau, B.; Schuckmann, L.V.; Thomas, S.; Rayner, J.E.; Spelman, L.; Wagner, G.; et al. Clinical utility of skin cancer and melanoma risk scores for population screening: TRoPICS study. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 1094–1098. [Google Scholar] [CrossRef]
- Trager, M.H.; Geskin, L.J.; Samie, F.H.; Liu, L. Biomarkers in melanoma and non-melanoma skin cancer prevention and risk stratification. Exp. Dermatol. 2022, 31, 4–12. [Google Scholar] [CrossRef]
- Thornton, J.; Chhabra, G.; Singh, C.K.; Guzmán-Pérez, G.; Shirley, C.A.; Ahmad, N. Mechanisms of Immunotherapy Resistance in Cutaneous Melanoma: Recognizing a Shapeshifter. Front. Oncol. 2022, 12, 880876. [Google Scholar] [CrossRef] [PubMed]
- Seretis, K.; Boptsi, E.; Boptsi, A.; Lykoudis, E.G. The impact of treatment delay on skin cancer in COVID-19 era: A case-control study. World J. Surg. Oncol. 2021, 19, 350. [Google Scholar] [CrossRef] [PubMed]
- Cives, M.; Mannavola, F.; Lospalluti, L.; Sergi, M.C.; Cazzato, G.; Filoni, E.; Cavallo, F.; Giudice, G.; Stucci, L.S.; Porta, C.; et al. Non-Melanoma Skin Cancers: Biological and Clinical Features. Int. J. Mol. Sci. 2020, 21, 5394. [Google Scholar] [CrossRef] [PubMed]
- Diaconeasa, A.; Boda, D.; Neagu, M.; Constantin, C.; Căruntu, C.; Vlădău, L.; Guţu, D. The role of confocal microscopy in the dermato-oncology practice. J. Med. Life 2011, 4, 63–74. [Google Scholar] [PubMed]
- Ghita, M.A.; Caruntu, C.; Rosca, A.E.; Kaleshi, H.; Caruntu, A.; Moraru, L.; Docea, A.O.; Zurac, S.; Boda, D.; Neagu, M.; et al. Reflectance confocal microscopy and dermoscopy for in vivo, non-invasive skin imaging of superficial basal cell carcinoma. Oncol. Lett. 2016, 11, 3019–3024. [Google Scholar] [CrossRef] [Green Version]
- Ilie, M.A.; Caruntu, C.; Lixandru, D.; Tampa, M.; Georgescu, S.-R.; Constantin, M.-M.; Constantin, C.; Neagu, M.; Zurac, S.A.; Boda, D. In vivo confocal laser scanning microscopy imaging of skin inflammation: Clinical applications and research directions. Exp. Ther. Med. 2019, 17, 1004–1011. [Google Scholar] [CrossRef] [Green Version]
- Van Herck, Y.; Antoranz, A.; Andhari, M.D.; Milli, G.; Bechter, O.; De Smet, F.; Bosisio, F.M. Multiplexed Immunohistochemistry and Digital Pathology as the Foundation for Next-Generation Pathology in Melanoma: Methodological Comparison and Future Clinical Applications. Front. Oncol. 2021, 11, 636681. [Google Scholar] [CrossRef]
- Tay, E.Y.-X.; Teoh, Y.-L.; Yeo, M.S.-W. Hedgehog Pathway Inhibitors and Their Utility in Basal Cell Carcinoma: A Comprehensive Review of Current Evidence. Dermatol. Ther. 2019, 9, 33–49. [Google Scholar] [CrossRef] [Green Version]
- Dobre, E.-G.; Constantin, C.; Costache, M.; Neagu, M. Interrogating Epigenome toward Personalized Approach in Cutaneous Melanoma. J. Pers. Med. 2021, 11, 901. [Google Scholar] [CrossRef]
- Dalle, S.; Mortier, L.; Corrie, P.; Lotem, M.; Board, R.; Arance, A.M.; Meiss, F.; Terheyden, P.; Gutzmer, R.; Buysse, B.; et al. Long-term real-world experience with ipilimumab and non-ipilimumab therapies in advanced melanoma: The IMAGE study. BMC Cancer 2021, 21, 642. [Google Scholar] [CrossRef]
- Reuben, A.; Spencer, C.N.; Prieto, P.A.; Gopalakrishnan, V.; Reddy, S.M.; Miller, J.P.; Mao, X.; De Macedo, M.P.; Chen, J.; Song, X.; et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med. 2017, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Tampa, M.; Georgescu, S.R.; Mitran, C.I.; Mitran, M.I.; Matei, C.; Scheau, C.; Constantin, C.; Neagu, M. Recent Advances in Signaling Pathways Comprehension as Carcinogenesis Triggers in Basal Cell Carcinoma. J. Clin. Med. 2020, 9, 3010. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Antona, C.; Taron, M. Pharmacogenomic biomarkers for personalized cancer treatment. J. Intern. Med. 2015, 277, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Maciejko, L.; Smalley, M.; Goldman, A. Cancer Immunotherapy and Personalized Medicine: Emerging Technologies and Biomarker-Based Approaches. J. Mol. Biomark. Diagn. 2017, 8, 350. [Google Scholar] [CrossRef]
- Kim, G.; Lee, S.K.; Suh, D.H.; Kim, K.; No, J.H.; Kim, Y.B.; Kim, H. Clinical evaluation of a droplet digital PCR assay for detecting POLE mutations and molecular classification of endometrial cancer. J. Gynecol. Oncol. 2022, 33, e15. [Google Scholar] [CrossRef]
- Villalba, M.; Exposito, F.; Pajares, M.J.; Sainz, C.; Redrado, M.; Remirez, A.; Wistuba, I.; Behrens, C.; Jantus-Lewintre, E.; Camps, C.; et al. TMPRSS4: A Novel Tumor Prognostic Indicator for the Stratification of Stage IA Tumors and a Liquid Biopsy Biomarker for NSCLC Patients. J. Clin. Med. 2019, 8, 2134. [Google Scholar] [CrossRef] [Green Version]
- Tsao, S.C.-H.; Weiss, J.; Hudson, C.; Christophi, C.; Cebon, J.; Behren, A.; Dobrovic, A. Monitoring response to therapy in melanoma by quantifying circulating tumour DNA with droplet digital PCR for BRAF and NRAS mutations. Sci. Rep. 2015, 5, 11198. [Google Scholar] [CrossRef]
- Demaree, B.; Weisgerber, D.; Dolatmoradi, A.; Hatori, M.; Abate, A.R. Direct quantification of EGFR variant allele frequency in cell-free DNA using a microfluidic-free digital droplet PCR assay. Methods Cell Biol. 2018, 148, 119–131. [Google Scholar] [CrossRef]
- Ito, T.; Kawashima, Y.; Fujikawa, T.; Honda, K.; Makabe, A.; Kitamura, K.; Tsutsumi, T. Rapid screening of copy number variations in STRC by droplet digital PCR in patients with mild-to-moderate hearing loss. Hum. Genome Var. 2019, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- van Ginkel, J.H.; Huibers, M.M.H.; van Es, R.J.J.; de Bree, R.; Willems, S.M. Droplet digital PCR for detection and quantification of circulating tumor DNA in plasma of head and neck cancer patients. BMC Cancer 2017, 17, 428. [Google Scholar] [CrossRef]
- van Zogchel, L.M.J.; Lak, N.S.M.; Verhagen, O.J.H.M.; Tissoudali, A.; Gussmalla Nuru, M.; Gelineau, N.U.; Zappeij-Kannengieter, L.; Javadi, A.; Zijtregtop, E.A.M.; Merks, J.H.M.; et al. Novel Circulating Hypermethylated RASSF1A ddPCR for Liquid Biopsies in Patients With Pediatric Solid Tumors. JCO Precis. Oncol. 2021, 5, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Crimi, S.; Falzone, L.; Gattuso, G.; Grillo, C.M.; Candido, S.; Bianchi, A.; Libra, M. Droplet Digital PCR Analysis of Liquid Biopsy Samples Unveils the Diagnostic Role of hsa-miR-133a-3p and hsa-miR-375-3p in Oral Cancer. Biology 2020, 9, 379. [Google Scholar] [CrossRef]
- De Paolis, E.; De Bonis, M.; Concolino, P.; Piermattei, A.; Fagotti, A.; Urbani, A.; Scambia, G.; Minucci, A.; Capoluongo, E. Droplet digital PCR for large genomic rearrangements detection: A promising strategy in tissue BRCA1 testing. Clin. Chim. Acta. 2021, 513, 17–24. [Google Scholar] [CrossRef]
- Ding, P.N.; Becker, T.; Bray, V.; Chua, W.; Ma, Y.; Xu, B.; Lynch, D.; de Souza, P.; Roberts, T. Plasma next generation sequencing and droplet digital PCR-based detection of epidermal growth factor receptor (EGFR) mutations in patients with advanced lung cancer treated with subsequent-line osimertinib. Thorac. Cancer 2019, 10, 1879–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta, M.; Roselló, S.; Sabater, L.; Ferrer, A.; Tarazona, N.; Roda, D.; Gambardella, V.; Alfaro-Cervelló, C.; Garcés-Albir, M.; Cervantes, A.; et al. Circulating Tumor DNA Detection by Digital-Droplet PCR in Pancreatic Ductal Adenocarcinoma: A Systematic Review. Cancers 2021, 13, 994. [Google Scholar] [CrossRef]
- Coccaro, N.; Tota, G.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Digital PCR: A Reliable Tool for Analyzing and Monitoring Hematologic Malignancies. Int. J. Mol. Sci. 2020, 21, 3141. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Hu, S.; Zhang, L.; Xin, J.; Sun, C.; Wang, L.; Ding, K.; Wang, B. Tumor circulome in the liquid biopsies for cancer diagnosis and prognosis. Theranostics 2020, 10, 4544–4556. [Google Scholar] [CrossRef]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: The potentiality of blood samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef]
- Tavano, F.; Gioffreda, D.; Valvano, M.R.; Palmieri, O.; Tardio, M.; Latiano, T.P.; Piepoli, A.; Maiello, E.; Pirozzi, F.; Andriulli, A. Droplet digital PCR quantification of miR-1290 as a circulating biomarker for pancreatic cancer. Sci. Rep. 2018, 8, 16389. [Google Scholar] [CrossRef]
- Del Re, M.; Vivaldi, C.; Rofi, E.; Vasile, E.; Miccoli, M.; Caparello, C.; d’Arienzo, P.D.; Fornaro, L.; Falcone, A.; Danesi, R. Early changes in plasma DNA levels of mutant KRAS as a sensitive marker of response to chemotherapy in pancreatic cancer. Sci. Rep. 2017, 7, 7931. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.J.G.; Hamilton, G.; Hurst, C.D.; Fraser, S.; Orange, C.; Knowles, M.A.; Jones, R.J.; Leung, H.Y.; Iwata, T. Monitoring of urothelial cancer disease status after treatment by digital droplet PCR liquid biopsy assays. Urol. Oncol. 2020, 38, e1–e737. [Google Scholar] [CrossRef] [PubMed]
- Parietti, M.; Marra, E.; Ribero, S.; Abate, S.O.; Francia di Celle, P.; Rudà, R.; Quaglino, P.; Fierro, M.T. Leptomeningeal dissemination as a first sign of progression in metastatic melanoma: A diagnostic lesson. Melanoma Res. 2022, 32, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Satyal, U.; Srivastava, A.; Abbosh, P.H. Urine Biopsy—Liquid Gold for Molecular Detection and Surveillance of Bladder Cancer. Front. Oncol. 2019, 9, 1266. [Google Scholar] [CrossRef] [Green Version]
- Sykes, P.J.; Neoh, S.H.; Brisco, M.J.; Hughes, E.; Condon, J.; Morley, A.A. Quantitation of targets for PCR by use of limiting dilution. Biotechniques 1992, 13, 444–449. [Google Scholar]
- Simmonds, P.; Balfe, P.; Peutherer, J.F.; Ludlam, C.A.; Bishop, J.O.; Brown, A.J. Human immunodeficiency virus-infected individuals contain provirus in small numbers of peripheral mononuclear cells and at low copy numbers. J. Virol. 1990, 64, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Ruano, G.; Kidd, K.K.; Stephens, J.C. Haplotype of multiple polymorphisms resolved by enzymatic amplification of single DNA molecules. Proc. Natl. Acad. Sci. USA 1990, 87, 6296–6300. [Google Scholar] [CrossRef] [Green Version]
- Brisco, M.J.; Condon, J.; Hughes, E.; Neoh, S.H.; Sykes, P.J.; Seshadri, R.; Toogood, I.; Waters, K.; Tauro, G.; Ekert, H. Outcome prediction in childhood acute lymphoblastic leukaemia by molecular quantification of residual disease at the end of induction. Lancet 1994, 343, 196–200. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [Green Version]
- Morley, A.A. Digital PCR: A brief history. Biomol. Detect. Quantif. 2014, 1, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Huggett, J.F.; Cowen, S.; Foy, C.A. Considerations for digital PCR as an accurate molecular diagnostic tool. Clin. Chem. 2015, 61, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solé, X.; Crous-Bou, M.; Cordero, D.; Olivares, D.; Guinó, E.; Sanz-Pamplona, R.; Rodriguez-Moranta, F.; Sanjuan, X.; de Oca, J.; Salazar, R.; et al. Discovery and validation of new potential biomarkers for early detection of colon cancer. PLoS ONE 2014, 9, e106748. [Google Scholar] [CrossRef]
- Arance, E.; Ramírez, V.; Rubio-Roldan, A.; Ocaña-Peinado, F.M.; Romero-Cachinero, C.; Jódar-Reyes, A.B.; Vazquez-Alonso, F.; Martinez-Gonzalez, L.J.; Alvarez-Cubero, M.J. Determination of Exosome Mitochondrial DNA as a Biomarker of Renal Cancer Aggressiveness. Cancers 2022, 14, 199. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.M.; Busby, E.; Garson, J.A.; Grant, P.R.; Nastouli, E.; Devonshire, A.S.; Whale, A.S. Digital PCR dynamic range is approaching that of real-time quantitative PCR. Biomol. Detect. Quantif. 2016, 10, 31–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corné, J.; Le Du, F.; Quillien, V.; Godey, F.; Robert, L.; Bourien, H.; Brunot, A.; Crouzet, L.; Perrin, C.; Lefeuvre-Plesse, C.; et al. Development of multiplex digital PCR assays for the detection of PIK3CA mutations in the plasma of metastatic breast cancer patients. Sci. Rep. 2021, 11, 17316. [Google Scholar] [CrossRef] [PubMed]
- García-Foncillas, J.; Alba, E.; Aranda, E.; Díaz-Rubio, E.; López-López, R.; Tabernero, J.; Vivancos, A. Incorporating BEAMing technology as a liquid biopsy into clinical practice for the management of colorectal cancer patients: An expert taskforce review. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 2943–2949. [Google Scholar] [CrossRef]
- Lee, H.; Lee, C.-J.; Kim, D.H.; Cho, C.-S.; Shin, W.; Han, K. High-accuracy quantitative principle of a new compact digital PCR equipment: Lab On An Array. Genomics Inform. 2021, 19, e34. [Google Scholar] [CrossRef]
- Wu, X.; Lee, Y.H.; Lu, T.K.; Yu, H. A Warm-start Digital CRISPR-based Method for the Quantitative Detection of Nucleic Acids. medRxiv 2021. [Google Scholar] [CrossRef]
- Manoj, P. Droplet digital PCR technology promises new applications and research areas. Mitochondrial DNA 2016, 27, 742–746. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Surcel, M. Testing Antigens, Antibodies, and Immune Cells in COVID-19 as a Public Health Topic-Experience and Outlines. Int. J. Environ. Res. Public Health 2021, 18, 13173. [Google Scholar] [CrossRef]
- Brink, B.G.; Meskas, J.; Brinkman, R.R. ddPCRclust: An R package and Shiny app for automated analysis of multiplexed ddPCR data. Bioinformatics 2018, 34, 2687–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Bell, A.D.; Usher, C.L.; McCarroll, S.A. Analyzing Copy Number Variation with Droplet Digital PCR. In Digital PCR: Methods and Protocols; Karlin-Neumann, G., Bizouarn, F., Eds.; Springer: New York, NY, USA, 2018; pp. 143–160. ISBN 978-1-4939-7778-9. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Nolan, T.; Pfaffl, M.W. Quantitative real-time RT-PCR--a perspective. J. Mol. Endocrinol. 2005, 34, 597–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Giménez, J.L.; Beltrán-García, J.; Romá-Mateo, C.; Seco-Cervera, M.; Pérez-Machado, G.; Mena-Mollá, S. Chapter 2–Epigenetic biomarkers for disease diagnosis. In Prognostic Epigenetics; Translational, Epigenetics; Sharma, S., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 15, pp. 21–44. [Google Scholar]
- Armbruster, D.A.; Pry, T. Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev. 2008, 29 (Suppl. S1), S49–S52. [Google Scholar]
- McEvoy, A.C.; Wood, B.A.; Ardakani, N.M.; Pereira, M.R.; Pearce, R.; Cowell, L.; Robinson, C.; Grieu-Iacopetta, F.; Spicer, A.J.; Amanuel, B.; et al. Droplet Digital PCR for Mutation Detection in Formalin-Fixed, Paraffin-Embedded Melanoma Tissues: A Comparison with Sanger Sequencing and Pyrosequencing. J. Mol. Diagn. 2018, 20, 240–252. [Google Scholar] [CrossRef] [Green Version]
- Deprez, L.; Corbisier, P.; Kortekaas, A.-M.; Mazoua, S.; Beaz Hidalgo, R.; Trapmann, S.; Emons, H. Validation of a digital PCR method for quantification of DNA copy number concentrations by using a certified reference material. Biomol. Detect. Quantif. 2016, 9, 29–39. [Google Scholar] [CrossRef]
- Huggett, J.F. The Digital MIQE Guidelines Update: Minimum Information for Publication of Quantitative Digital PCR Experiments for 2020. Clin. Chem. 2020, 66, 1012–1029. [Google Scholar] [CrossRef]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The digital MIQE guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef]
- Rowlands, V.; Rutkowski, A.J.; Meuser, E.; Carr, T.H.; Harrington, E.A.; Barrett, J.C. Optimisation of robust singleplex and multiplex droplet digital PCR assays for high confidence mutation detection in circulating tumour DNA. Sci. Rep. 2019, 9, 12620. [Google Scholar] [CrossRef] [Green Version]
- Park, G.; Park, J.K.; Son, D.-S.; Shin, S.-H.; Kim, Y.J.; Jeon, H.-J.; Lee, J.; Park, W.-Y.; Lee, K.H.; Park, D. Utility of targeted deep sequencing for detecting circulating tumor DNA in pancreatic cancer patients. Sci. Rep. 2018, 8, 11631. [Google Scholar] [CrossRef] [PubMed]
- Bohers, E.; Viailly, P.-J.; Jardin, F. cfDNA Sequencing: Technological Approaches and Bioinformatic Issues. Pharmaceuticals 2021, 14, 596. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R. Next-Generation Sequencing in High-Sensitive Detection of Mutations in Tumors: Challenges, Advances, and Applications. J. Mol. Diagn. 2020, 22, 994–1007. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Dobre, E.-G.; Neagu, M. Droplet Digital PCR: An Emerging Technology for Cutaneous Melanoma Detection and Monitoring. Biol. Life Sci. Forum 2021, 7, 20. [Google Scholar] [CrossRef]
- Vanni, I.; Tanda, E.T.; Spagnolo, F.; Andreotti, V.; Bruno, W.; Ghiorzo, P. The Current State of Molecular Testing in the BRAF-Mutated Melanoma Landscape. Front. Mol. Biosci. 2020, 7, 113. [Google Scholar] [CrossRef]
- Chen, M.-L.; Wang, S.-H.; Wei, J.C.-C.; Yip, H.-T.; Hung, Y.-M.; Chang, R. The Impact of Human Papillomavirus Infection on Skin Cancer: A Population-Based Cohort Study. Oncologist 2021, 26, e473–e483. [Google Scholar] [CrossRef]
- Patel, A.S.; Karagas, M.R.; Perry, A.E.; Nelson, H.H. Exposure profiles and human papillomavirus infection in skin cancer: An analysis of 25 genus beta-types in a population-based study. J. Investig. Dermatol. 2008, 128, 2888–2893. [Google Scholar] [CrossRef] [Green Version]
- Boda, D.; Docea, A.O.; Calina, D.; Ilie, M.A.; Caruntu, C.; Zurac, S.; Neagu, M.; Constantin, C.; Branisteanu, D.E.; Voiculescu, V.; et al. Human papilloma virus: Apprehending the link with carcinogenesis and unveiling new research avenues (Review). Int. J. Oncol. 2018, 52, 637–655. [Google Scholar] [CrossRef] [Green Version]
- Rollison, D.E.; Amorrortu, R.P.; Zhao, Y.; Messina, J.L.; Schell, M.J.; Fenske, N.A.; Cherpelis, B.S.; Giuliano, A.R.; Sondak, V.K.; Pawlita, M.; et al. Cutaneous Human Papillomaviruses and the Risk of Keratinocyte Carcinomas. Cancer Res. 2021, 81, 4628–4638. [Google Scholar] [CrossRef]
- Schiavetto, C.M.; de Abreu, P.M.; von Zeidler, S.V.; de Jesus, L.M.; Carvalho, R.S.; Cirino, M.T.; Carloni, A.C.; Oliveira, C.; Scapulatempo-Neto, C.; de Almeida, G.C.; et al. Human Papillomavirus DNA Detection by Droplet Digital PCR in Formalin-Fixed Paraffin-Embedded Tumor Tissue from Oropharyngeal Squamous Cell Carcinoma Patients. Mol. Diagn. Ther. 2021, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Melis, J.P.M.; Luijten, M.; Mullenders, L.H.F.; van Steeg, H. The role of XPC: Implications in cancer and oxidative DNA damage. Mutat. Res. 2011, 728, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkouteren, B.J.A.; Cosgun, B.; Reinders, M.G.H.C.; Kessler, P.A.W.K.; Vermeulen, R.J.; Klaassens, M.; Lambrechts, S.; van Rheenen, J.R.; van Geel, M.; Vreeburg, M.; et al. A guideline for the clinical management of basal cell naevus syndrome (Gorlin–Goltz syndrome). Br. J. Dermatol. 2022, 186, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Nutsathapana, N.; Bunnag, T.; Chaowalit, P.; Supsrisunjai, C. Basal Cell Nevus Syndrome caused by a new splice site mutation in PTCH1. Med. Sci. Discov. 2021, 8, 289–290. [Google Scholar] [CrossRef]
- Reinders, M.G.H.C. The Hedgehog Pathway in Basal Cell Carcinoma: Target for Diagnostics and Therapy; Maastricht University: Maastricht, The Netherlands, 2019. [Google Scholar]
- Aoude, L.G.; Wadt, K.A.W.; Pritchard, A.L.; Hayward, N.K. Genetics of familial melanoma: 20 years after CDKN2A. Pigment Cell Melanoma Res. 2015, 28, 148–160. [Google Scholar] [CrossRef]
- Dunnett-Kane, V.; Burkitt-Wright, E.; Blackhall, F.H.; Malliri, A.; Evans, D.G.; Lindsay, C.R. Germline and sporadic cancers driven by the RAS pathway: Parallels and contrasts. Ann. Oncol. 2020, 31, 873–883. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Malicherova, B.; Burjanivova, T.; Grendar, M.; Minarikova, E.; Bobrovska, M.; Vanova, B.; Jasek, K.; Jezkova, E.; Kapinova, A.; Antosova, M.; et al. Droplet digital PCR for detection of BRAF V600E mutation in formalin-fixed, paraffin-embedded melanoma tissues: A comparison with Cobas(®) 4800, Sanger sequencing, and allele-specific PCR. Am. J. Transl. Res. 2018, 10, 3773–3781. [Google Scholar]
- Ghanadan, A.; Yousefi, T.; Kamyab-Hesari, K.; Azhari, V.; Nasimi, M. Prevalence and Main Determinants of BRAF V600E Mutation in Dysplastic and Congenital Nevi. Iran. J. Pathol. 2021, 16, 51–56. [Google Scholar] [CrossRef]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Berghe, A.S.; Cobzac, G.; Dindelegan, G.; Șenilă, S.C.; Baican, C.I.; Solomon, C.M.; Rogojan, L.; Leucuța, D.C.; Drugan, T.C.; Bolboacă, S.D. Risk factors for positive sentinel lymph node, lymphatic or hematogenous dissemination over time in patients with cutaneous melanoma. Exp. Ther. Med. 2021, 22, 730. [Google Scholar] [CrossRef] [PubMed]
- Skin Cancer in Australia, Summary—Australian Institute of Health and Welfare. Available online: https://www.aihw.gov.au/reports/cancer/skin-cancer-in-australia/summary-1 (accessed on 11 July 2022).
- Mays, M.P.; Martin, R.C.G.; Burton, A.; Ginter, B.; Edwards, M.J.; Reintgen, D.S.; Ross, M.I.; Urist, M.M.; Stromberg, A.J.; McMasters, K.M.; et al. Should all patients with melanoma between 1 and 2 mm Breslow thickness undergo sentinel lymph node biopsy? Cancer 2010, 116, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Falk Delgado, A.; Zommorodi, S.; Falk Delgado, A. Sentinel Lymph Node Biopsy and Complete Lymph Node Dissection for Melanoma. Curr. Oncol. Rep. 2019, 21, 54. [Google Scholar] [CrossRef] [Green Version]
- Seynnaeve, B.; Lee, S.; Borah, S.; Park, Y.; Pappo, A.; Kirkwood, J.M.; Bahrami, A. Genetic and Epigenetic Alterations of TERT Are Associated with Inferior Outcome in Adolescent and Young Adult Patients with Melanoma. Sci. Rep. 2017, 7, 45704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.D.; Leão, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolónio, J.D.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Motaparthi, K.; Kim, J.; Andea, A.A.; Missall, T.A.; Novoa, R.A.; Vidal, C.I.; Fung, M.A.; Emanuel, P.O. TERT and TERT promoter in melanocytic neoplasms: Current concepts in pathogenesis, diagnosis, and prognosis. J. Cutan. Pathol. 2020, 47, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Salgado, C.; Roelse, C.; Nell, R.; Gruis, N.; van Doorn, R.; van der Velden, P. Interplay between TERT promoter mutations and methylation culminates in chromatin accessibility and TERT expression. PLoS ONE 2020, 15, e0231418. [Google Scholar] [CrossRef] [Green Version]
- Griewank, K.G.; Murali, R.; Puig-Butille, J.A.; Schilling, B.; Livingstone, E.; Potrony, M.; Carrera, C.; Schimming, T.; Möller, I.; Schwamborn, M.; et al. TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J. Natl. Cancer Inst. 2014, 106, dju246. [Google Scholar] [CrossRef]
- Heppt, M.V.; Siepmann, T.; Engel, J.; Schubert-Fritschle, G.; Eckel, R.; Mirlach, L.; Kirchner, T.; Jung, A.; Gesierich, A.; Ruzicka, T.; et al. Prognostic significance of BRAF and NRAS mutations in melanoma: A German study from routine care. BMC Cancer 2017, 17, 536. [Google Scholar] [CrossRef]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef]
- Garcia-Alvarez, A.; Ortiz, C.; Muñoz-Couselo, E. Current Perspectives and Novel Strategies of NRAS-Mutant Melanoma. Onco. Targets. Ther. 2021, 14, 3709–3719. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, M.; Giunta, E.F.; Tortora, M.; Curvietto, M.; Attademo, L.; Bosso, D.; Cardalesi, C.; Rosanova, M.; De Placido, P.; Pietroluongo, E.; et al. BRAF Gene and Melanoma: Back to the Future. Int. J. Mol. Sci. 2021, 22, 3474. [Google Scholar] [CrossRef] [PubMed]
- Rabbie, R.; Ferguson, P.; Wong, K.; Couturier, D.-L.; Moran, U.; Turner, C.; Emanuel, P.; Haas, K.; Saunus, J.M.; Davidson, M.R.; et al. The mutational landscape of melanoma brain metastases presenting as the first visceral site of recurrence. Br. J. Cancer 2020, 124, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Forgione, L.; Carotenuto, M.; De Luca, A.; Ascierto, P.A.; Botti, G.; Normanno, N. Circulating Tumor DNA Testing Opens New Perspectives in Melanoma Management. Cancers 2020, 12, 2914. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef]
- Chang, G.A.; Wiggins, J.M.; Corless, B.C.; Syeda, M.M.; Tadepalli, J.S.; Blake, S.; Fleming, N.; Darvishian, F.; Pavlick, A.; Berman, R.; et al. TERT, BRAF, and NRAS Mutational Heterogeneity between Paired Primary and Metastatic Melanoma Tumors. J. Investig. Dermatol. 2020, 140, 1609–1618.e7. [Google Scholar] [CrossRef]
- Nikolouzakis, T.K.; Falzone, L.; Lasithiotakis, K.; Krüger-Krasagakis, S.; Kalogeraki, A.; Sifaki, M.; Spandidos, D.A.; Chrysos, E.; Tsatsakis, A.; Tsiaoussis, J. Current and Future Trends in Molecular Biomarkers for Diagnostic, Prognostic, and Predictive Purposes in Non-Melanoma Skin Cancer. J. Clin. Med. 2020, 9, 2868. [Google Scholar] [CrossRef]
- Hu, P.; Ma, L.; Wu, Z.; Zheng, G.; Li, J. Expression of miR-34a in basal cell carcinoma patients and its relationship with prognosis. J. BUON. 2019, 24, 1283–1288. [Google Scholar]
- Cañueto, J.; Cardeñoso-Álvarez, E.; García-Hernández, J.L.; Galindo-Villardón, P.; Vicente-Galindo, P.; Vicente-Villardón, J.L.; Alonso-López, D.; De Las Rivas, J.; Valero, J.; Moyano-Sanz, E.; et al. MicroRNA (miR)-203 and miR-205 expression patterns identify subgroups of prognosis in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2017, 177, 168–178. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Dobre, E.-G.; Dinescu, S.; Costache, M. Connecting the missing dots: Ncrnas as critical regulators of therapeutic susceptibility in breast cancer. Cancers 2020, 12, 2698. [Google Scholar] [CrossRef] [PubMed]
- Laprovitera, N.; Riefolo, M.; Porcellini, E.; Durante, G.; Garajova, I.; Vasuri, F.; Aigelsreiter, A.; Dandachi, N.; Benvenuto, G.; Agostinis, F.; et al. MicroRNA expression profiling with a droplet digital PCR assay enables molecular diagnosis and prognosis of cancers of unknown primary. Mol. Oncol. 2021, 15, 2732–2751. [Google Scholar] [CrossRef] [PubMed]
- Likhacheva, A.; Awan, M.; Barker, C.A.; Bhatnagar, A.; Bradfield, L.; Brady, M.S.; Buzurovic, I.; Geiger, J.L.; Parvathaneni, U.; Zaky, S.; et al. Definitive and Postoperative Radiation Therapy for Basal and Squamous Cell Cancers of the Skin: Executive Summary of an American Society for Radiation Oncology Clinical Practice Guideline. Pract. Radiat. Oncol. 2020, 10, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Switzer, B.; Puzanov, I.; Skitzki, J.J.; Hamad, L.; Ernstoff, M.S. Managing Metastatic Melanoma in 2022: A Clinical Review. JCO Oncol. Pract. 2022, 18, 335–351. [Google Scholar] [CrossRef]
- Jennings, L.; Schmults, C.D. Management of high-risk cutaneous squamous cell carcinoma. J. Clin. Aesthet. Dermatol. 2010, 3, 39–48. [Google Scholar] [PubMed]
- Hong, A.M.; Fogarty, G.B.; Dolven-Jacobsen, K.; Burmeister, B.H.; Lo, S.N.; Haydu, L.E.; Vardy, J.L.; Nowak, A.K.; Dhillon, H.M.; Ahmed, T.; et al. Adjuvant Whole-Brain Radiation Therapy Compared With Observation After Local Treatment of Melanoma Brain Metastases: A Multicenter, Randomized Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 3132–3141. [Google Scholar] [CrossRef]
- Stonesifer, C.J.; Djavid, A.R.; Grimes, J.M.; Khaleel, A.E.; Soliman, Y.S.; Maisel-Campbell, A.; Garcia-Saleem, T.J.; Geskin, L.J.; Carvajal, R.D. Immune Checkpoint Inhibition in Non-Melanoma Skin Cancer: A Review of Current Evidence. Front. Oncol. 2021, 11, 734354. [Google Scholar] [CrossRef]
- Sun, Q.; Atzmony, L.; Zaki, T.; Peng, A.; Sugarman, J.; Choate, K.A. Clues to primary vismodegib resistance lie in histology and genetics. J. Clin. Pathol. 2020, 73, 678–680. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N. Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Ascierto, P.A.; Basset-Seguin, N.; Dréno, B.; Garbe, C.; Gutzmer, R.; Hauschild, A.; Krattinger, R.; Lear, J.T.; Malvehy, J.; et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: A joint expert opinion. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1944–1956. [Google Scholar] [CrossRef]
- Sarkisian, S.; Davar, D. MEK inhibitors for the treatment of NRAS mutant melanoma. Drug Des. Devel. Ther. 2018, 12, 2553–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niezgoda, A.; Niezgoda, P.; Czajkowski, R. Novel Approaches to Treatment of Advanced Melanoma: A Review on Targeted Therapy and Immunotherapy. Biomed Res. Int. 2015, 2015, 851387. [Google Scholar] [CrossRef] [Green Version]
- Alberti, A.; Bossi, P. Immunotherapy for Cutaneous Squamous Cell Carcinoma: Results and Perspectives. Front. Oncol. 2022, 11, 727027. [Google Scholar] [CrossRef]
- Asher, N.; Ben-Betzalel, G.; Lev-Ari, S.; Shapira-Frommer, R.; Steinberg-Silman, Y.; Gochman, N.; Schachter, J.; Meirson, T.; Markel, G. Real World Outcomes of Ipilimumab and Nivolumab in Patients with Metastatic Melanoma. Cancers 2020, 12, 2329. [Google Scholar] [CrossRef] [PubMed]
- Grob, J.J.; Mendoza, R.G.; Basset-Seguin, N.; Vornicova, O.; Schachter, J.; Joshi, A.; Meyer, N.; Grange, F.; Piulats, J.M.; Bauman, J.; et al. Pembrolizumab for recurrent/metastatic cutaneous squamous cell carcinoma (cSCC): Efficacy and safety results from the phase II KEYNOTE-629 study. Ann. Oncol. 2019, 30, v908. [Google Scholar] [CrossRef]
- Stratigos, A.J.; Sekulic, A.; Peris, K.; Bechter, O.; Dutriaux, C.; Kaatz, M.; Lewis, K.D.; Basset-Seguin, N.; Chang, A.L.S.; Dalle, S.; et al. LBA47 Primary analysis of phase II results for cemiplimab in patients (pts) with locally advanced basal cell carcinoma (laBCC) who progress on or are intolerant to hedgehog inhibitors (HHIs). Ann. Oncol. 2020, 31, S1175–S1176. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, L.; Song, J.; Wang, G.; Li, P.; Li, W.; Luo, P.; Sun, X.; Wu, J.; Liu, Y.; et al. Liquid biopsy at the frontier of detection, prognosis and progression monitoring in colorectal cancer. Mol. Cancer 2022, 21, 86. [Google Scholar] [CrossRef] [PubMed]
- Michela, B. Liquid Biopsy: A Family of Possible Diagnostic Tools. Diagnostics 2021, 11, 1391. [Google Scholar] [CrossRef]
- Olmedillas-López, S.; García-Arranz, M.; García-Olmo, D. Current and Emerging Applications of Droplet Digital PCR in Oncology. Mol. Diagn. Ther. 2017, 21, 493–510. [Google Scholar] [CrossRef]
- Burjanivova, T.; Malicherova, B.; Grendar, M.; Minarikova, E.; Dusenka, R.; Vanova, B.; Bobrovska, M.; Pecova, T.; Homola, I.; Lasabova, Z.; et al. Detection of BRAFV600E Mutation in Melanoma Patients by Digital PCR of Circulating DNA. Genet. Test. Mol. Biomark. 2019, 23, 241–245. [Google Scholar] [CrossRef]
- Pinzani, P.; D’Argenio, V.; Re, M.D.; Pellegrini, C.; Cucchiara, F.; Salvianti, F.; Galbiati, S. Updates on liquid biopsy: Current trends and future perspectives for clinical application in solid tumors. Clin. Chem. Lab. Med. 2021, 59, 1181–1200. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. Comparison of methods for the isolation of cell-free DNA from cell culture supernatant. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2020, 42, 1010428320916314. [Google Scholar] [CrossRef]
- Lim, S.Y.; Lee, J.H.; Diefenbach, R.J.; Kefford, R.F.; Rizos, H. Liquid biomarkers in melanoma: Detection and discovery. Mol. Cancer 2018, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Grob, J.-J.; Nathan, P.; Ribas, A.; Robert, C.; Schadendorf, D.; Lane, S.R.; Mak, C.; Legenne, P.; Flaherty, K.T.; et al. Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: A pooled analysis of individual patient data from randomised trials. Lancet. Oncol. 2016, 17, 1743–1754. [Google Scholar] [CrossRef]
- Diem, S.; Kasenda, B.; Martin-Liberal, J.; Lee, A.; Chauhan, D.; Gore, M.; Larkin, J. Prognostic score for patients with advanced melanoma treated with ipilimumab. Eur. J. Cancer 2015, 51, 2785–2791. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Manda, G.; Margaritescu, I. Biomarkers of metastatic melanoma. Biomark. Med. 2009, 3, 71–89. [Google Scholar] [CrossRef]
- Tang, H.; Kong, Y.; Si, L.; Cui, C.; Sheng, X.; Chi, Z.; Dai, J.; Yu, S.; Ma, M.; Wu, X.; et al. Clinical significance of BRAF(V600E) mutation in circulating tumor DNA in Chinese patients with melanoma. Oncol. Lett. 2018, 15, 1839–1844. [Google Scholar] [CrossRef] [Green Version]
- Shinozaki, M.; O’Day, S.J.; Kitago, M.; Amersi, F.; Kuo, C.; Kim, J.; Wang, H.-J.; Hoon, D.S.B. Utility of circulating B-RAF DNA mutation in serum for monitoring melanoma patients receiving biochemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 2068–2074. [Google Scholar] [CrossRef] [Green Version]
- Syeda, M.M.; Wiggins, J.M.; Corless, B.C.; Long, G.V.; Flaherty, K.T.; Schadendorf, D.; Nathan, P.D.; Robert, C.; Ribas, A.; Davies, M.A.; et al. Circulating tumour DNA in patients with advanced melanoma treated with dabrafenib or dabrafenib plus trametinib: A clinical validation study. Lancet Oncol. 2021, 22, 370–380. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Fernández-Landázuri, S.; Rodríguez, C.; Zárate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martín-Algarra, S.; González, A. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Marsavela, G.; Lee, J.; Calapre, L.; Wong, S.Q.; Pereira, M.R.; McEvoy, A.C.; Reid, A.L.; Robinson, C.; Warburton, L.; Abed, A.; et al. Circulating Tumor DNA Predicts Outcome from First-, but not Second-line Treatment and Identifies Melanoma Patients Who May Benefit from Combination Immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 5926–5933. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Saw, R.P.; Thompson, J.F.; Lo, S.; Spillane, A.J.; Shannon, K.F.; Stretch, J.R.; Howle, J.; Menzies, A.M.; Carlino, M.S.; et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Gouda, M.A.; Polivka, J.; Huang, H.J.; Treskova, I.; Pivovarcikova, K.; Fikrle, T.; Woznica, V.; Dustin, D.J.; Call, S.G.; Meric-Bernstam, F.; et al. Ultrasensitive detection of BRAF mutations in circulating tumor DNA of non-metastatic melanoma. ESMO Open 2022, 7, 100357. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.J.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Seremet, T.; Jansen, Y.; Planken, S.; Njimi, H.; Delaunoy, M.; El Housni, H.; Awada, G.; Schwarze, J.K.; Keyaerts, M.; Everaert, H.; et al. Undetectable circulating tumor DNA (ctDNA) levels correlate with favorable outcome in metastatic melanoma patients treated with anti-PD1 therapy. J. Transl. Med. 2019, 17, 303. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wang, Q.; Dong, Q.; Zhan, L.; Zhang, J. How to differentiate pseudoprogression from true progression in cancer patients treated with immunotherapy. Am. J. Cancer Res. 2019, 9, 1546–1553. [Google Scholar]
- Lee, J.H.; Long, G.V.; Menzies, A.M.; Lo, S.; Guminski, A.; Whitbourne, K.; Peranec, M.; Scolyer, R.; Kefford, R.F.; Rizos, H.; et al. Association Between Circulating Tumor DNA and Pseudoprogression in Patients With Metastatic Melanoma Treated With Anti-Programmed Cell Death 1 Antibodies. JAMA Oncol. 2018, 4, 717–721. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Menzies, A.M.; Carlino, M.S.; McEvoy, A.C.; Sandhu, S.; Weppler, A.M.; Diefenbach, R.J.; Dawson, S.-J.; Kefford, R.F.; Millward, M.J.; et al. Longitudinal Monitoring of ctDNA in Patients with Melanoma and Brain Metastases Treated with Immune Checkpoint Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 4064–4071. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Elemento, O. Analyzing DNA Methylation Patterns During Tumor Evolution. Methods Mol. Biol. 2018, 1711, 27–53. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Lee, J.H.; Rizos, H. Methylated circulating tumor DNA as a biomarker in cutaneous melanoma. Melanoma Manag. 2020, 7, MMT46. [Google Scholar] [CrossRef] [PubMed]
- Aleotti, V.; Catoni, C.; Poggiana, C.; Rosato, A.; Facchinetti, A.; Scaini, M.C. Methylation Markers in Cutaneous Melanoma: Unravelling the Potential Utility of Their Tracking by Liquid Biopsy. Cancers 2021, 13, 6217. [Google Scholar] [CrossRef]
- Mori, T.; O’Day, S.J.; Umetani, N.; Martinez, S.R.; Kitago, M.; Koyanagi, K.; Kuo, C.; Takeshima, T.-L.; Milford, R.; Wang, H.-J.; et al. Predictive utility of circulating methylated DNA in serum of melanoma patients receiving biochemotherapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 9351–9358. [Google Scholar] [CrossRef] [PubMed]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenetics 2017, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Sigalotti, L.; Fratta, E.; Bidoli, E.; Covre, A.; Parisi, G.; Colizzi, F.; Coral, S.; Massarut, S.; Kirkwood, J.M.; Maio, M. Methylation levels of the “long interspersed nucleotide element-1” repetitive sequences predict survival of melanoma patients. J. Transl. Med. 2011, 9, 78. [Google Scholar] [CrossRef] [Green Version]
- Haselmann, V.; Hedtke, M.; Neumaier, M. Liquid Profiling for Cancer Patient Stratification in Precision Medicine- Current Status and Challenges for Successful Implementation in Standard Care. Diagnostics 2022, 12, 748. [Google Scholar] [CrossRef]
- Avanzini, S.; Kurtz, D.M.; Chabon, J.J.; Moding, E.J.; Hori, S.S.; Gambhir, S.S.; Alizadeh, A.A.; Diehn, M.; Reiter, J.G. A mathematical model of ctDNA shedding predicts tumor detection size. Sci. Adv. 2020, 6, eabc4308. [Google Scholar] [CrossRef]
- Keller, L.; Belloum, Y.; Wikman, H.; Pantel, K. Clinical relevance of blood-based ctDNA analysis: Mutation detection and beyond. Br. J. Cancer 2021, 124, 345–358. [Google Scholar] [CrossRef]
- Otandault, A.; Abraham, J.-D.; Al Amir Dache, Z.; Khalyfa, A.; Jariel-Encontre, I.; Forné, T.; Prévostel, C.; Chouaib, S.; Gozal, D.; Thierry, A.R. Hypoxia differently modulates the release of mitochondrial and nuclear DNA. Br. J. Cancer 2020, 122, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Abbosh, C.; Birkbak, N.J.; Swanton, C. Early stage NSCLC—challenges to implementing ctDNA-based screening and MRD detection. Nat. Rev. Clin. Oncol. 2018, 15, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Forschner, A.; Weißgraeber, S.; Hadaschik, D.; Schulze, M.; Kopp, M.; Kelkenberg, S.; Sinnberg, T.; Garbe, C.; Biskup, S.; Battke, F. Circulating Tumor DNA Correlates with Outcome in Metastatic Melanoma Treated by BRAF and MEK Inhibitors—Results of a Prospective Biomarker Study. Onco. Targets. Ther. 2020, 13, 5017–5032. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Liu, T.; Qiao, L.; Gao, M.; Li, J. Decreased serum microRNA-206 level predicts unfavorable prognosis in patients with melanoma. Int. J. Clin. Exp. Pathol. 2015, 8, 3097–3103. [Google Scholar]
- Xu, Y.; Brenn, T.; Brown, E.R.S.; Doherty, V.; Melton, D.W. Differential expression of microRNAs during melanoma progression: miR-200c, miR-205 and miR-211 are downregulated in melanoma and act as tumour suppressors. Br. J. Cancer 2012, 106, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.-H.; Zhou, F.; Shi, C.; Xiang, T.; Zhou, C.-K.; Wang, Q.-Q.; Jiang, Y.-S.; Gao, S.-F. miRNA-221 promotes cutaneous squamous cell carcinoma progression by targeting PTEN. Cell. Mol. Biol. Lett. 2019, 24, 9. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.; Constantin, C.; Cretoiu, S.M.; Zurac, S. miRNAs in the Diagnosis and Prognosis of Skin Cancer. Front. cell Dev. Biol. 2020, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Bryzgunova, O.; Konoshenko, M.; Zaporozhchenko, I.; Yakovlev, A.; Laktionov, P. Isolation of Cell-Free miRNA from Biological Fluids: Influencing Factors and Methods. Diagnostics 2021, 11, 865. [Google Scholar] [CrossRef]
- Faraldi, M.; Gomarasca, M.; Banfi, G.; Lombardi, G. Free Circulating miRNAs Measurement in Clinical Settings: The Still Unsolved Issue of the Normalization. Adv. Clin. Chem. 2018, 87, 113–139. [Google Scholar] [CrossRef]
- Fogli, S.; Polini, B.; Carpi, S.; Pardini, B.; Naccarati, A.; Dubbini, N.; Lanza, M.; Breschi, M.C.; Romanini, A.; Nieri, P. Identification of plasma microRNAs as new potential biomarkers with high diagnostic power in human cutaneous melanoma. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2017, 39, 1010428317701646. [Google Scholar] [CrossRef] [Green Version]
- Jayawardana, K.; Schramm, S.-J.; Tembe, V.; Mueller, S.; Thompson, J.F.; Scolyer, R.A.; Mann, G.J.; Yang, J. Identification, Review, and Systematic Cross-Validation of microRNA Prognostic Signatures in Metastatic Melanoma. J. Investig. Dermatol. 2016, 136, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Fattore, L.; Mancini, R.; Acunzo, M.; Romano, G.; Laganà, A.; Pisanu, M.E.; Malpicci, D.; Madonna, G.; Mallardo, D.; Capone, M.; et al. miR-579-3p controls melanoma progression and resistance to target therapy. Proc. Natl. Acad. Sci. USA 2016, 113, E5005–E5013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakar, K.; Rodrίguez, C.I.; Jayanthy, A.S.; Mikheil, D.M.; Bhasker, A.I.; Perera, R.J.; Setaluri, V. Role of miR-214 in regulation of β-catenin and the malignant phenotype of melanoma. Mol. Carcinog. 2019, 58, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poenitzsch Strong, A.M.; Setaluri, V.; Spiegelman, V.S. MicroRNA-340 as a modulator of RAS-RAF-MAPK signaling in melanoma. Arch. Biochem. Biophys. 2014, 563, 118–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirillo, P.D.R.; Margiotti, K.; Mesoraca, A.; Giorlandino, C. Quantification of circulating microRNAs by droplet digital PCR for cancer detection. BMC Res. Notes 2020, 13, 351. [Google Scholar] [CrossRef]
- van Zweeden, A.A.; Opperman, R.C.M.; Honeywell, R.J.; Peters, G.J.; Verheul, H.M.W.; van der Vliet, H.J.; Poel, D. The prognostic impact of circulating miRNAs in patients with advanced esophagogastric cancer during palliative chemotherapy. Cancer Treat. Res. Commun. 2021, 27, 100371. [Google Scholar] [CrossRef]
- Shoji, Y.; Bustos, M.A.; Gross, R.; Hoon, D.S.B. Recent Developments of Circulating Tumor Cell Analysis for Monitoring Cutaneous Melanoma Patients. Cancers 2022, 14, 859. [Google Scholar] [CrossRef]
- Morosin, T.; Ashford, B.; Ranson, M.; Gupta, R.; Clark, J.; Iyer, N.G.; Spring, K. Circulating tumour cells in regionally metastatic cutaneous squamous cell carcinoma: A pilot study. Oncotarget 2016, 7, 47111–47115. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Fu, W.; Zhang, W.; Li, P. High Number of Circulating Tumor Cells Predicts Poor Survival of Cutaneous Melanoma Patients in China. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Lei, K.F. A Review on Microdevices for Isolating Circulating Tumor Cells. Micromachines 2020, 11, 531. [Google Scholar] [CrossRef]
- Reid, A.L.; Freeman, J.B.; Millward, M.; Ziman, M.; Gray, E.S. Detection of BRAF-V600E and V600K in melanoma circulating tumour cells by droplet digital PCR. Clin. Biochem. 2015, 48, 999–1002. [Google Scholar] [CrossRef]
- Denis, J.A.; Patroni, A.; Guillerm, E.; Pépin, D.; Benali-Furet, N.; Wechsler, J.; Manceau, G.; Bernard, M.; Coulet, F.; Larsen, A.K.; et al. Droplet digital PCR of circulating tumor cells from colorectal cancer patients can predict KRAS mutations before surgery. Mol. Oncol. 2016, 10, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- Hoshimoto, S.; Faries, M.B.; Morton, D.L.; Shingai, T.; Kuo, C.; Wang, H.-J.; Elashoff, R.; Mozzillo, N.; Kelley, M.C.; Thompson, J.F.; et al. Assessment of prognostic circulating tumor cells in a phase III trial of adjuvant immunotherapy after complete resection of stage IV melanoma. Ann. Surg. 2012, 255, 357–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanojevic, M.; Grant, M.; Nazarian, J.; Knoblach, S.; Panditharatna, E.; Panchapakesan, K.; Gress, R.E.; Chakrabarty, J.H.; Wiliams, K.M. Simultaneous Detection of Circulating Tumor Antigens in Acute Leukemia after HSCT. Biol. Blood Marrow Transplant. 2020, 26, S123. [Google Scholar] [CrossRef]
- Hong, X.; Sullivan, R.J.; Kalinich, M.; Kwan, T.T.; Giobbie-Hurder, A.; Pan, S.; LiCausi, J.A.; Milner, J.D.; Nieman, L.T.; Wittner, B.S.; et al. Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 2467–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, D.; Wang, D.; Sanchez-Garcia, J.F.; Cusano, M.P.; Miller, W.; Cai, T.; Scheuenpflug, J.; Feng, Z. Fit-for-purpose quantitative liquid biopsy based droplet digital PCR assay development for detection of programmed cell death ligand-1 (PD-L1) RNA expression in PAXgene blood samples. PLoS ONE 2021, 16, e0250849. [Google Scholar] [CrossRef]
- Strati, A.; Zavridou, M.; Economopoulou, P.; Gkolfinopoulos, S.; Psyrri, A.; Lianidou, E. Development and Analytical Validation of a Reverse Transcription Droplet Digital PCR (RT-ddPCR) Assay for PD-L1 Transcripts in Circulating Tumor Cells. Clin. Chem. 2021, 67, 642–652. [Google Scholar] [CrossRef]
- Tan, Z.; Yue, C.; Ji, S.; Zhao, C.; Jia, R.; Zhang, Y.; Liu, R.; Li, D.; Yu, Q.; Li, P.; et al. Assessment of PD-L1 Expression on Circulating Tumor Cells for Predicting Clinical Outcomes in Patients with Cancer Receiving PD-1/PD-L1 Blockade Therapies. Oncologist 2021, 26, e2227–e2238. [Google Scholar] [CrossRef]
- Cayrefourcq, L.; Alix-Panabières, C. CTCs as Liquid Biopsy: Where Are We Now? In Molecular Medicine; Nalbantoglu, S., Amri, H., Eds.; IntechOpen: Rijeka, Croatia, 2019. [Google Scholar]
- Study of Circulating Tumor Cells Before and After Treatment in Patients With Metastatic Melanoma-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01573494 (accessed on 24 April 2022).
- Ex Vivo Expansion of Circulating Tumor Cells as a Model for Cancer Predictive Pharmacology-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03797053?cond=circulating+tumor+cells.melanoma&draw=2&rank=5 (accessed on 24 April 2022).
- Heat Shock Protein (HSP) 70 to Quantify and Characterize Circulating Tumor Cells-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04628806?cond=circulating+tumor+cells.melanoma&draw=2&rank=6 (accessed on 24 April 2022).
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Tamura, T.; Yoshioka, Y.; Sakamoto, S.; Ichikawa, T.; Ochiya, T. Extracellular vesicles as a promising biomarker resource in liquid biopsy for cancer. Extracell. Vesicles Circ. Nucleic Acids 2021, 2, 148–174. [Google Scholar] [CrossRef]
- Willms, E.; Cabañas, C.; Mäger, I.; Wood, M.J.A.; Vader, P. Extracellular Vesicle Heterogeneity: Subpopulations, Isolation Techniques, and Diverse Functions in Cancer Progression. Front. Immunol. 2018, 9, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Kakarla, R.; Hur, J.; Kim, Y.J.; Kim, J.; Chwae, Y.-J. Apoptotic cell-derived exosomes: Messages from dying cells. Exp. Mol. Med. 2020, 52, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanasi, I.; Adamo, A.; Kamga, P.T.; Bazzoni, R.; Krampera, M. High-throughput analysis and functional interpretation of extracellular vesicle content in hematological malignancies. Comput. Struct. Biotechnol. J. 2020, 18, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Caby, M.-P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 2005, 17, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Beretti, F.; Zavatti, M.; Casciaro, F.; Comitini, G.; Franchi, F.; Barbieri, V.; La Sala, G.B.; Maraldi, T. Amniotic fluid stem cell exosomes: Therapeutic perspective. Biofactors 2018, 44, 158–167. [Google Scholar] [CrossRef]
- Street, J.M.; Koritzinsky, E.H.; Glispie, D.M.; Star, R.A.; Yuen, P.S.T. Urine Exosomes: An Emerging Trove of Biomarkers. Adv. Clin. Chem. 2017, 78, 103–122. [Google Scholar] [CrossRef]
- Sjoqvist, S.; Otake, K.; Hirozane, Y. Analysis of Cerebrospinal Fluid Extracellular Vesicles by Proximity Extension Assay: A Comparative Study of Four Isolation Kits. Int. J. Mol. Sci. 2020, 21, 9425. [Google Scholar] [CrossRef]
- Kupsco, A.; Prada, D.; Valvi, D.; Hu, L.; Petersen, M.S.; Coull, B.; Grandjean, P.; Weihe, P.; Baccarelli, A.A. Human milk extracellular vesicle miRNA expression and associations with maternal characteristics in a population-based cohort from the Faroe Islands. Sci. Rep. 2021, 11, 5840. [Google Scholar] [CrossRef]
- Chiabotto, G.; Gai, C.; Deregibus, M.C.; Camussi, G. Salivary Extracellular Vesicle-Associated exRNA as Cancer Biomarker. Cancers 2019, 11, 891. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Yoshida-Court, K.; Solley, T.N.; Mikkelson, M.; Yeung, C.L.A.; Nick, A.; Lu, K.; Klopp, A.H. Extracellular vesicles derived from ascitic fluid enhance growth and migration of ovarian cancer cells. Sci. Rep. 2021, 11, 9149. [Google Scholar] [CrossRef] [PubMed]
- Pieragostino, D.; Lanuti, P.; Cicalini, I.; Cufaro, M.C.; Ciccocioppo, F.; Ronci, M.; Simeone, P.; Onofrj, M.; van der Pol, E.; Fontana, A.; et al. Proteomics characterization of extracellular vesicles sorted by flow cytometry reveals a disease-specific molecular cross-talk from cerebrospinal fluid and tears in multiple sclerosis. J. Proteom. 2019, 204, 103403. [Google Scholar] [CrossRef]
- Höög, J.L.; Lötvall, J. Diversity of extracellular vesicles in human ejaculates revealed by cryo-electron microscopy. J. Extracell. Vesicles 2015, 4, 28680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dlugolecka, M.; Szymanski, J.; Zareba, L.; Homoncik, Z.; Domagala-Kulawik, J.; Polubiec-Kownacka, M.; Czystowska-Kuzmicz, M. Characterization of Extracellular Vesicles from Bronchoalveolar Lavage Fluid and Plasma of Patients with Lung Lesions Using Fluorescence Nanoparticle Tracking Analysis. Cells 2021, 10, 3473. [Google Scholar] [CrossRef]
- Staubach, S.; Bauer, F.N.; Tertel, T.; Börger, V.; Stambouli, O.; Salzig, D.; Giebel, B. Scaled preparation of extracellular vesicles from conditioned media. Adv. Drug Deliv. Rev. 2021, 177, 113940. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Sun, J.; Wang, X.; Hu, T.; Ma, Y.; Kong, C.; Piao, H.; Yu, T.; Zhang, G. Exosomes: A Promising Avenue for the Diagnosis of Breast Cancer. Technol. Cancer Res. Treat. 2019, 18, 1533033818821421. [Google Scholar] [CrossRef] [Green Version]
- Vesiclepedia: Home-Extracellular vesicles database. Available online: http://microvesicles.org/ (accessed on 22 April 2022).
- Ramirez, M.I.; Amorim, M.G.; Gadelha, C.; Milic, I.; Welsh, J.A.; Freitas, V.M.; Nawaz, M.; Akbar, N.; Couch, Y.; Makin, L.; et al. Technical challenges of working with extracellular vesicles. Nanoscale 2018, 10, 881–906. [Google Scholar] [CrossRef] [Green Version]
- Soekmadji, C.; Li, B.; Huang, Y.; Wang, H.; An, T.; Liu, C.; Pan, W.; Chen, J.; Cheung, L.; Falcon-Perez, J.M.; et al. The future of Extracellular Vesicles as Theranostics-an ISEV meeting report. J. Extracell. Vesicles 2020, 9, 1809766. [Google Scholar] [CrossRef]
- Zocco, D.; Bernardi, S.; Novelli, M.; Astrua, C.; Fava, P.; Zarovni, N.; Carpi, F.M.; Bianciardi, L.; Malavenda, O.; Quaglino, P.; et al. Isolation of extracellular vesicles improves the detection of mutant DNA from plasma of metastatic melanoma patients. Sci. Rep. 2020, 10, 15745. [Google Scholar] [CrossRef]
- Tangella, L.P.; Clark, M.E.; Gray, E.S. Resistance mechanisms to targeted therapy in BRAF-mutant melanoma-A mini review. Biochim. Biophys. Acta-Gen. Subj. 2021, 1865, 129736. [Google Scholar] [CrossRef]
- Clark, M.E.; Rizos, H.; Pereira, M.R.; McEvoy, A.C.; Marsavela, G.; Calapre, L.; Meehan, K.; Ruhen, O.; Khattak, M.A.; Meniawy, T.M.; et al. Detection of BRAF splicing variants in plasma-derived cell-free nucleic acids and extracellular vesicles of melanoma patients failing targeted therapy therapies. Oncotarget 2020, 11, 4016–4027. [Google Scholar] [CrossRef]
- Yap, S.A.; Münster-Wandowski, A.; Nonnenmacher, A.; Keilholz, U.; Liebs, S. Analysis of cancer-related mutations in extracellular vesicles RNA by Droplet DigitalTM PCR. Biotechniques 2020, 69, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Lone, S.N.; Nisar, S.; Masoodi, T.; Singh, M.; Rizwan, A.; Hashem, S.; El-Rifai, W.; Bedognetti, D.; Batra, S.K.; Haris, M.; et al. Liquid biopsy: A step closer to transform diagnosis, prognosis and future of cancer treatments. Mol. Cancer 2022, 21, 79. [Google Scholar] [CrossRef] [PubMed]
- Murad, A.M.; Carneiro, J.G.; Casali-da-Rocha, J.C. A single institution experience with droplet digital polymerase chain reaction (dd-PCR) liquid biopsy (LB) for therapeutic decision in advanced solid tumors. J. Clin. Oncol. 2021, 39, 3038. [Google Scholar] [CrossRef]
- Lee, J.H.J.; Long, G.V.; Menzies, A.M.; Guminski, A.D.; Kefford, R.; Rizos, H.; Carlino, M.S. Analysis of circulating tumor DNA (ctDNA) in pseudoprogression in anti-PD1 treated metastatic melanoma (MM). J. Clin. Oncol. 2017, 35, 9546. [Google Scholar] [CrossRef]
- Lee, J.H.J.; Menzies, A.M.; Carlino, M.S.; Kefford, R.; Scolyer, R.A.; Long, G.V.; Rizos, H. Circulating tumor DNA (ctDNA) in metastatic melanoma (MM) patients (pts) with brain metastases (mets). J. Clin. Oncol. 2019, 37, 9581. [Google Scholar] [CrossRef]
- Kumar, R.; Angelini, S.; Snellman, E.; Hemminki, K. BRAF Mutations Are Common Somatic Events in Melanocytic Nevi. J. Investig. Dermatol. 2004, 122, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.-S.; Park, C.H.; Lee, S.; Park, H.S. Clinicopathological parameters for circulating tumor DNA shedding in surgically resected non-small cell lung cancer with EGFR or KRAS mutation. PLoS ONE 2020, 15, e0230622. [Google Scholar] [CrossRef] [Green Version]
- Schuh, S.; Ruini, C.; Perwein, M.K.E.; Daxenberger, F.; Gust, C.; Sattler, E.C.; Welzel, J. Line-Field Confocal Optical Coherence Tomography: A New Tool for the Differentiation between Nevi and Melanomas? Cancers 2022, 14, 1140. [Google Scholar] [CrossRef]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.; Kefford, R.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF Mutation Status in Circulating-Free DNA and Tumor and Association with Clinical Outcome across Four BRAFi and MEKi Clinical Trials. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, R.; Lee, J.; Chandler, D.; Wang, Y.; Pflueger, C.; Long, G.; Scolyer, R.; Carlino, M.; Menzies, A.; Kefford, R.; et al. Hypermethylation of Circulating Free DNA in Cutaneous Melanoma. Appl. Sci. 2019, 9, 5074. [Google Scholar] [CrossRef] [Green Version]
- Khattak, M.A.; Reid, A.; Freeman, J.; Pereira, M.; McEvoy, A.; Lo, J.; Frank, M.H.; Meniawy, T.; Didan, A.; Spencer, I.; et al. PD-L1 Expression on Circulating Tumor Cells May Be Predictive of Response to Pembrolizumab in Advanced Melanoma: Results from a Pilot Study. Oncologist 2019, 25, e520–e527. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Technology | Assay | Sensitivity | Specificity | LoD | Type of Alterations | Strengths | Limitations | Ref. |
|---|---|---|---|---|---|---|---|---|
| Real-time PCR | AS-PCR | 1% | 98% | 0.001% | Know point mutations (SNVs, Fusions, Indels CNVs) | Ease of design and execution; High sensitivity and specificity of detection with fluorescent hydrolysis probes; No need for informatics expert support. | Detects only known genomic variants in limited genomic regions; Reduced multiplexing capability; Quantitation requires standard curve using appropriate positive controls. | [75,76] |
| MS-PCR | 0.62% | 89–100% | 0.1% | Known methylation sites | Ease of design and execution; Increased sensitivity when analyzing small quantities of methylated DNA; No need for informatics expert support. | Detects only specific CpG islands. | [76] | |
| ddPCR | 0.001–0.1% | 100% | 0.005% | Know point mutations (SNVs, Fusions, Indels, CNVs) | Absolute quantitation possible because of scanning and Poisson-based counting of droplets; No need for a standard curve for quantitation; Short turnaround time; No need for informatics expert support. | Unsuitable for mutation screening and identification of novel variants; Reduced multiplexing Capability. | [68,75,76] | |
| NGS | WGS | 5–10% | 80–99.9% | 5–10% | Genome-wide CNVs, DNA methylation studies | Prior knowledge of mutations not required; Genome-wide profiling; Identification of specific cancer signatures. Pathogenic gene screening; Detection of CNVs, fusion genes, rearrangements, neoantigens and TMB. | Extensive bioinformatics support; Variable sensitivity and specificity (increase depth leads to higher costs); Long turnaround time; Costly and not appropriate for patient longitudinal monitoring. | [76] |
| WES | 5% | 80–95.6% | 5% | Coding regions, gene promoters, intron-exon junctions, non-coding DNA of miRNA genes | ||||
| TargetedNGS gene panels | 0.01–0.1% | 99.6% | 2–5% | Know point mutations | Increased sensitivity and specificity compared to WES/WGS; Produces a smaller and more manageable data set compared to untargeted approaches, making analysis easier. | Less comprehensive than WES/WGS; amplicon methods based on multiplex PCR. | [74] | |
| Sanger sequencing | 15–20% | 100% | 20–25% | Know point mutations | Provides sequence information and determines whether a point mutation or small deletion/duplication is present. | Low sensitivity; Low discovery power; Costly and laborious. | [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobre, E.-G.; Constantin, C.; Neagu, M. Skin Cancer Research Goes Digital: Looking for Biomarkers within the Droplets. J. Pers. Med. 2022, 12, 1136. https://doi.org/10.3390/jpm12071136
Dobre E-G, Constantin C, Neagu M. Skin Cancer Research Goes Digital: Looking for Biomarkers within the Droplets. Journal of Personalized Medicine. 2022; 12(7):1136. https://doi.org/10.3390/jpm12071136
Chicago/Turabian StyleDobre, Elena-Georgiana, Carolina Constantin, and Monica Neagu. 2022. "Skin Cancer Research Goes Digital: Looking for Biomarkers within the Droplets" Journal of Personalized Medicine 12, no. 7: 1136. https://doi.org/10.3390/jpm12071136
APA StyleDobre, E.-G., Constantin, C., & Neagu, M. (2022). Skin Cancer Research Goes Digital: Looking for Biomarkers within the Droplets. Journal of Personalized Medicine, 12(7), 1136. https://doi.org/10.3390/jpm12071136