
The Role of Sphingomyelin and Ceramide in Motor Neuron Diseases
 , and
, and
Abstract
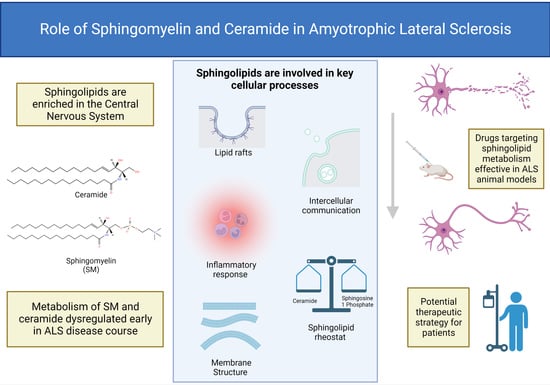
:
1. Introduction
2. Lipid Dysregulation in MNDs
2.1. Dyslipidaemia in ALS
2.2. Hyperlipidaemia in SBMA
2.3. Dysregulated Fatty Acid Metabolism in SMA
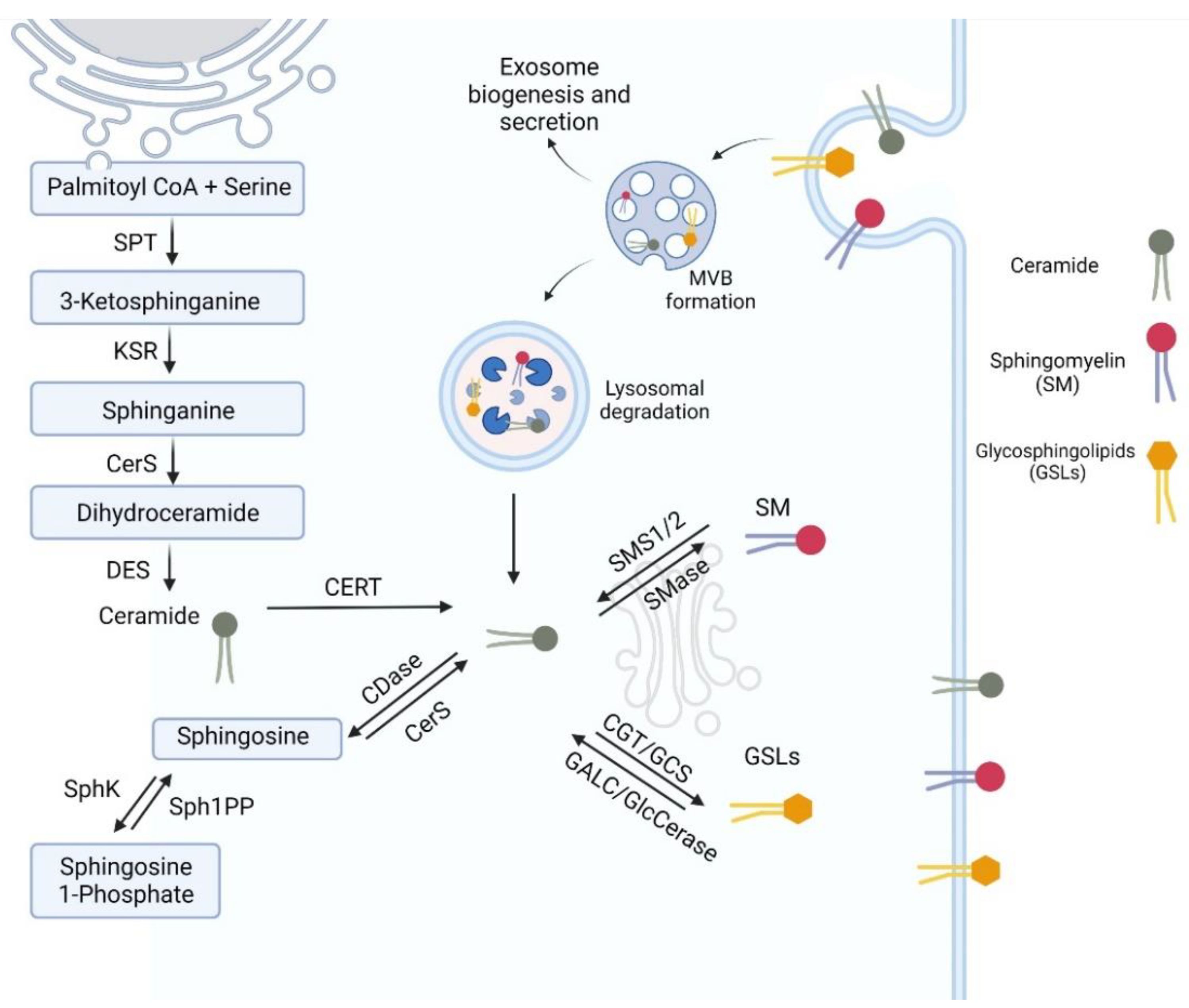
3. Sphingolipid Synthesis
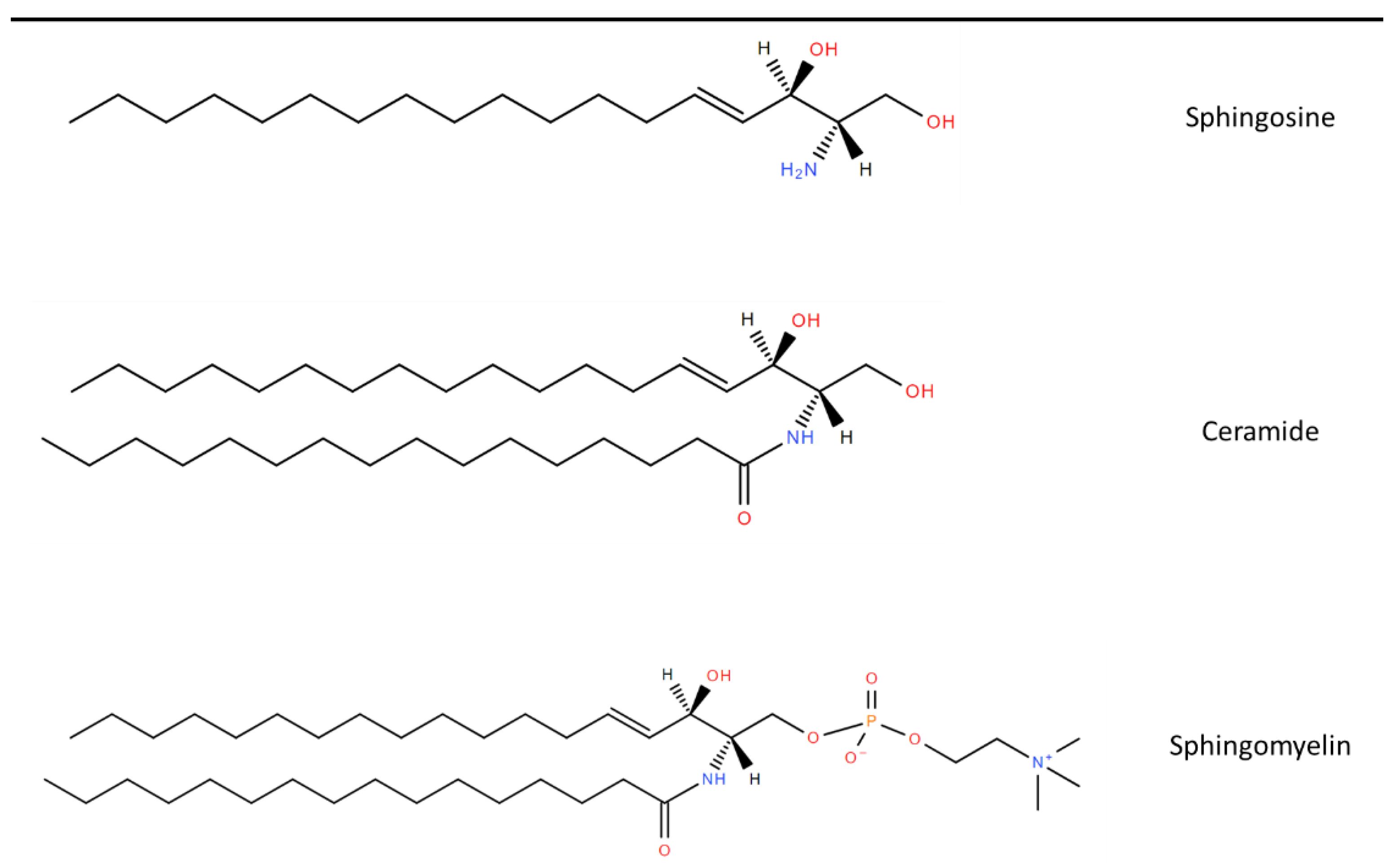
3.1. Ceramide Metabolism
3.2. Metabolism of Complex Sphingolipids
4. Biological Function of Ceramide and Sphingomyelin
5. Role of Sphingolipids in MNDs
6. Lipidomic Studies in MNDs
7. Potential Therapeutics Targeting Sphingolipid Metabolism
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zuccaro, E.; Piol, D.; Basso, M.; Pennuto, M. Motor Neuron Diseases and Neuroprotective Peptides: A Closer Look to Neurons. Front. Aging Neurosci. 2021, 13, 723871. [Google Scholar] [CrossRef] [PubMed]
- Riku, Y.; Atsuta, N.; Yoshida, M.; Tatsumi, S.; Iwasaki, Y.; Mimuro, M.; Watanabe, H.; Ito, M.; Senda, J.; Nakamura, R.; et al. Differential motor neuron involvement in progressive muscular atrophy: A comparative study with amyotrophic lateral sclerosis. BMJ Open 2014, 4, e005213. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, G.; Duddy, W.; Haffey, S.; Morrison, K.; Donaghy, C.; Duguez, S. Epidemiology and survival trends of motor neurone disease in Northern Ireland from 2015 to 2019. Eur. J. Neurol. 2022, 29, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Connolly, O.; Le Gall, L.; McCluskey, G.; Donaghy, C.G.; Duddy, W.J.; Duguez, S. A Systematic Review of Genotype-Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS. J. Pers. Med. 2020, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy—A literature review. Orphanet. J. Rare Dis. 2017, 12, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Schorling, D.C.; Pechmann, A.; Kirschner, J. Advances in Treatment of Spinal Muscular Atrophy—New Phenotypes, New Challenges, New Implications for Care. J. Neuromuscul. Dis. 2020, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef]
- Malek, E.G.; Salameh, J.S.; Makki, A. Kennedy’s disease: An under-recognized motor neuron disorder. Acta Neurol. Belg. 2020, 120, 1289–1295. [Google Scholar] [CrossRef]
- Pradat, P.-F., on behalf of the French Kennedy’s Disease Writing Group; Bernard, E.; Corcia, P.; Couratier, P.; Jublanc, C.; Querin, G.; Panzini, C.M.; Salachas, F.; Vial, C.; Wahbi, K.; et al. The French national protocol for Kennedy’s disease (SBMA): Consensus diagnostic and management recommendations. Orphanet. J. Rare Dis. 2020, 15, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parboosingh, J.S.; Figlewicz, D.A.; Krizus, A.; Meininger, V.; Azad, N.A.; Newman, D.S.; Rouleau, G.A. Spinobulbar muscular atrophy can mimic ALS: The importance of genetic testing in male patients with atypical ALS. Neurology 1997, 49, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, A.; Fischbeck, K.H.; Pennuto, M.; Fratta, P.; Katsuno, M. Disease mechanism, biomarker and therapeutics for spinal and bulbar muscular atrophy (SBMA). J. Neurol. Neurosurg. Psychiatry 2020, 91, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.; Hussain, G.; Dupuis, L.; Loeffler, J.; Henriques, A. A plural role for lipids in motor neuron diseases: Energy, signaling and structure. Front. Cell. Neurosci. 2014, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Pujol-Lereis, L.M. Alteration of Sphingolipids in Biofluids: Implications for Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3564. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Croixmarie, V.; Bouscary, A.; Mosbach, A.; Keime, C.; Boursier-Neyret, C.; Walter, B.; Spedding, M.; Loeffler, J.-P. Sphingolipid Metabolism Is Dysregulated at Transcriptomic and Metabolic Levels in the Spinal Cord of an Animal Model of Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 10, 433. [Google Scholar] [CrossRef]
- Cutler, R.G.; Pedersen, W.A.; Camandola, S.; Rothstein, J.D.; Mattson, M.P. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. 2002, 52, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Vargas, M.; Robinson, K.M.; Cassina, P.; Díaz-Amarilla, P.J.; Hagen, T.M.; Radi, R.; Barbeito, L.; Beckman, J.S. Mitochondrial superoxide production and nuclear factor erythroid 2-related factor 2 activation in p75 neurotrophin receptor-induced motor neuron apoptosis. J. Neurosci. 2007, 27, 7777–7785. [Google Scholar] [CrossRef] [PubMed]
- Steyn, F.; Ioannides, Z.; Van Eijk, R.P.; Heggie, S.; Thorpe, K.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; Berg, L.H.V.D.; et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef]
- Fayemendy, P.; Marin, B.; Labrunie, A.; Boirie, Y.; Walrand, S.; Achamrah, N.; Coëffier, M.; Preux, P.-M.; Lautrette, G.; Desport, J.-C.; et al. Hypermetabolism is a reality in amyotrophic lateral sclerosis compared to healthy subjects. J. Neurol. Sci. 2021, 420, 117257. [Google Scholar] [CrossRef]
- Ferri, A.; Coccurello, R. What is “Hyper” in the ALS Hypermetabolism? Mediat. Inflamm 2017, 2017, 7821672. [Google Scholar] [CrossRef]
- Reich-Slotky, R.; Andrews, J.; Cheng, B.; Buchsbaum, R.; Levy, D.; Kaufmann, P.; Thompson, J.L.P. Body mass index (BMI) as predictor of ALSFRS-R score decline in ALS patients. Amyotroph Lateral Scler. Front. Degener. 2013, 14, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Deng, J.; Jaffa, M.; Cudkowicz, M.E.; Wills, A.M. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 2011, 44, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Corcia, P.; Fergani, A.; De Aguilar, J.-G.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.-J.; Lacomblez, L.; Loeffler, J.-P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.; Talbot, K.; Turner, M.R. Higher blood high density lipoprotein and apolipoprotein A1 levels are associated with reduced risk of developing amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2022, 93, 75–81. [Google Scholar] [CrossRef]
- Zinman, L.; Sadeghi, R.; Gawel, M.; Patton, D.; Kiss, A. Are statin medications safe in patients with ALS? Amyotroph Lateral Scler. 2008, 9, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Mariosa, D.; Kamel, F.; Bellocco, R.; Ronnevi, L.; Almqvist, C.; Larsson, H.; Ye, W.; Fang, F. Antidiabetics, statins and the risk of amyotrophic lateral sclerosis. Eur. J. Neurol. 2020, 27, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Ji, H. Medications on hypertension, hyperlipidemia, diabetes, and risk of amyotrophic lateral sclerosis: A systematic review and meta-analysis. Neurol. Sci. 2022, 43, 5189–5199. [Google Scholar] [CrossRef]
- Weisskopf, M.G.; Levy, J.; Dickerson, A.S.; Paganoni, S.; Leventer-Roberts, M. Statin Medications and Amyotrophic Lateral Sclerosis Incidence and Mortality. Am. J. Epidemiol. 2022, 191, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-M.; Cheng, Y.-F.; Jiang, Z.; Duan, Q.-Q.; Yang, T.-M.; Shang, H.-F.; Chen, Y.-P. Predictors of survival in patients with amyotrophic lateral sclerosis: A large meta-analysis. EBioMedicine 2021, 74, 103732. [Google Scholar] [CrossRef]
- Francini-Pesenti, F.; Querin, G.; Martini, C.; Mareso, S.; Sacerdoti, D. Prevalence of metabolic syndrome and non-alcoholic fatty liver disease in a cohort of italian patients with spinal-bulbar muscular atrophy. Acta Myol. 2018, 37, 204–209. [Google Scholar] [PubMed]
- Rosenbohm, A.; Hirsch, S.; Volk, A.E.; Grehl, T.; Grosskreutz, J.; Hanisch, F.; Herrmann, A.; Kollewe, K.; Kress, W.; Meyer, T.; et al. The metabolic and endocrine characteristics in spinal and bulbar muscular atrophy. J. Neurol. 2018, 265, 1026–1036. [Google Scholar] [CrossRef]
- Singh, R.; Artaza, J.N.; Taylor, W.E.; Braga, M.; Yuan, X.; Gonzalez-Cadavid, N.F.; Bhasin, S. Testosterone inhibits adipogenic differentiation in 3T3-L1 cells: Nuclear translocation of androgen receptor complex with beta-catenin and T-cell factor 4 may bypass canonical Wnt signaling to down-regulate adipogenic transcription factors. Endocrinology 2006, 147, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Zitzmann, M.; Gromoll, J.; von Eckardstein, A.; Nieschlag, E. The CAG repeat polymorphism in the androgen receptor gene modulates body fat mass and serum concentrations of leptin and insulin in men. Diabetologia 2003, 46, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.; Gray, P.B.; Eisenberg, D.T.; Ellison, P.; Sorenson, M.D. Androgen receptor CAG repeats and body composition among Ariaal men. Int. J. Androl. 2009, 32, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, A.; Milioto, C.; Parodi, S.; Armirotti, A.; Borgia, D.; Pellegrini, M.; Urciuolo, A.; Molon, S.; Morbidoni, V.; Marabita, M.; et al. Glycolytic-to-oxidative fiber-type switch and mTOR signaling activation are early-onset features of SBMA muscle modified by high-fat diet. Acta Neuropathol. 2016, 132, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Deguise, M.; Chehade, L.; Kothary, R. Metabolic Dysfunction in Spinal Muscular Atrophy. Int. J. Mol. Sci. 2021, 22, 5913. [Google Scholar] [CrossRef]
- Davis, R.H.; Miller, E.A.; Zhang, R.Z.; Swoboda, K.J. Responses to Fasting and Glucose Loading in a Cohort of Well Children with Spinal Muscular Atrophy Type, I.I. J. Pediatr. 2015, 167, 1362. [Google Scholar] [CrossRef] [PubMed]
- Deguise, M.; Baranello, G.; Mastella, C.; Beauvais, A.; Michaud, J.; Leone, A.; De Amicis, R.; Battezzati, A.; Dunham, C.; Selby, K.; et al. Abnormal fatty acid metabolism is a core component of spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 1519–1532. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An Overview of Sphingolipid Metabolism: From Synthesis to Breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [PubMed]
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. J. Lipid Res. 2018, 59, 1325–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, D.C.; Aguilera-Albesa, S.; Pujol, A. Ceramide signalling in inherited and multifactorial brain metabolic diseases. Neurobiol. Dis. 2020, 143, 105014. [Google Scholar] [CrossRef] [PubMed]
- Fanani, M.L.; Maggio, B. The many faces (and phases) of ceramide and sphingomyelin I—Single lipids. Biophys Rev. 2017, 9, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Sud, M.; Cotter, D.; Subramaniam, S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007, 35, 606. [Google Scholar] [CrossRef] [PubMed]
- Slotte, J.P. Biological functions of sphingomyelins. Prog. Lipid Res. 2013, 52, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Kumagai, K.; Tomishige, N.; Kawano, M. CERT and intracellular trafficking of ceramide. Biochim. Biophys. Acta 2007, 1771, 644–653. [Google Scholar] [CrossRef]
- Young, S.A.; Mina, J.G.; Denny, P.W.; Smith, T.K. Sphingolipid and ceramide homeostasis: Potential therapeutic targets. Biochem. Res. Int. 2012, 2012, 248135. [Google Scholar] [CrossRef] [PubMed]
- Pavoine, C.; Pecker, F. Sphingomyelinases: Their regulation and roles in cardiovascular pathophysiology. Cardiovasc. Res. 2009, 82, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.D.; Cheng, Y.; Hansen, G.; Hertervig, E.; Liu, J.J.; Syk, I.; Sjostrom, H.; Nilsson, A. Purification, localization, and expression of human intestinal alkaline sphingomyelinase. J. Lipid Res. 2003, 44, 1241–1250. [Google Scholar] [CrossRef]
- Xiang, H.; Jin, S.; Tan, F.; Xu, Y.; Lu, Y.; Wu, T. Physiological functions and therapeutic applications of neutral sphingomyelinase and acid sphingomyelinase. Biomed Pharmacother. 2021, 139, 111610. [Google Scholar] [CrossRef]
- Tallon, C.; Picciolini, S.; Yoo, S.; Thomas, A.G.; Pal, A.; Alt, J.; Carlomagno, C.; Gualerzi, A.; Rais, R.; Haughey, N.J.; et al. Inhibition of neutral sphingomyelinase 2 reduces extracellular vesicle release from neurons, oligodendrocytes, and activated microglial cells following acute brain injury. Biochem. Pharmacol. 2021, 194, 114796. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, G.; Capasso, S.; Sticco, L.; Russo, D. Glycosphingolipids: Synthesis and functions. FEBS J. 2013, 280, 6338–6353. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, H.J.; Quiroga, R.; Ferrari, M.L. Cellular and molecular biology of glycosphingolipid glycosylation. J. Neurochem. 2011, 117, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Reza, S.; Ugorski, M.; Suchański, J. Glucosylceramide and galactosylceramide, small glycosphingolipids with significant impact on health and disease. Glycobiology 2021, 31, 1416–1434. [Google Scholar] [CrossRef] [PubMed]
- Schulze, H.; Sandhoff, K. Lysosomal lipid storage diseases. Cold Spring Harb. Perspect. Biol. 2011, 3, a004804. [Google Scholar] [CrossRef] [PubMed]
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar] [CrossRef] [PubMed]
- García-Arribas, A.B.; Alonso, A.; Goñi, F.M. Cholesterol interactions with ceramide and sphingomyelin. Chem. Phys. Lipids 2016, 199, 26–34. [Google Scholar] [CrossRef]
- Gupta, A.K.; Rudney, H. Plasma membrane sphingomyelin and the regulation of HMG-CoA reductase activity and cholesterol biosynthesis in cell cultures. J. Lipid Res. 1991, 32, 125–136. [Google Scholar] [CrossRef]
- Moll, T.; Marshall, J.N.G.; Soni, N.; Zhang, S.; Cooper-Knock, J.; Shaw, P.J. Membrane lipid raft homeostasis is directly linked to neurodegeneration. Essays Biochem. 2021, 65, 999–1011. [Google Scholar]
- Bieberich, E. Sphingolipids and lipid rafts: Novel concepts and methods of analysis. Chem Phys Lipids. 2018, 216, 114–131. [Google Scholar] [CrossRef]
- Engberg, O.; Lin, K.; Hautala, V.; Slotte, J.P.; Nyholm, T.K.M. Sphingomyelin Acyl Chains Influence the Formation of Sphingomyelin- and Cholesterol-Enriched Domains. Biophys. J. 2020, 119, 913–923. [Google Scholar] [CrossRef]
- Levental, I.; Levental, K.R.; Heberle, F.A. Lipid Rafts: Controversies Resolved, Mysteries Remain. Trends Cell Biol. 2020, 30, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Levental, I.; Wang, H.Y. Membrane domains beyond the reach of microscopy. J. Lipid Res. 2020, 61, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, Q.; Kakuda, S.; London, E. Nanodomains can persist at physiologic temperature in plasma membrane vesicles and be modulated by altering cell lipids. J. Lipid Res. 2020, 61, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Adame, P.L.; Meza, U.; Rodríguez-Menchaca, A.A.; Sánchez-Armass, S.; Ruiz-García, J.; Gomez, E. Determination of the size of lipid rafts studied through single-molecule FRET simulations. Biophys J. 2021, 120, 2287–2295. [Google Scholar] [CrossRef] [PubMed]
- Young, M.M.; Kester, M.; Wang, H.G. Sphingolipids: Regulators of crosstalk between apoptosis and autophagy. J. Lipid Res. 2013, 54, 5–19. [Google Scholar] [CrossRef]
- Sawada, M.; Kiyono, T.; Nakashima, S.; Shinoda, J.; Naganawa, T.; Hara, S.; Iwama, T.; Sakai, N. Molecular mechanisms of TNF-alpha-induced ceramide formation in human glioma cells: P53-mediated oxidant stress-dependent and -independent pathways. Cell Death Differ. 2004, 11, 997–1008. [Google Scholar] [CrossRef]
- Ariga, T.; Jarvis, W.D.; Yu, R.K. Role of sphingolipid-mediated cell death in neurodegenerative diseases. J. Lipid Res. 1998, 39, 1–16. [Google Scholar] [CrossRef]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, J.S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- Satoi, H.; Tomimoto, H.; Ohtani, R.; Kitano, T.; Kondo, T.; Watanabe, M.; Oka, N.; Akiguchi, I.; Furuya, S.; Hirabayashi, Y.; et al. Astroglial expression of ceramide in Alzheimer’s disease brains: A role during neuronal apoptosis. Neuroscience 2005, 130, 657–666. [Google Scholar] [CrossRef]
- Lee, J.T.; Xu, J.; Lee, J.M.; Ku, G.; Han, X.; Yang, D.I.; Chen, S.; Hsu, C.Y. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J. Cell Biol. 2004, 164, 123–131. [Google Scholar] [CrossRef]
- Brann, A.B.; Tcherpakov, M.; Williams, I.M.; Futerman, A.H.; Fainzilber, M. Nerve growth factor-induced p75-mediated death of cultured hippocampal neurons is age-dependent and transduced through ceramide generated by neutral sphingomyelinase. J. Biol. Chem. 2002, 277, 9812–9818. [Google Scholar] [CrossRef] [Green Version]
- Obinata, H.; Hla, T. Sphingosine 1-phosphate and inflammation. Int. Immunol. 2019, 31, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Anakor, E.; Le Gall, L.; Dumonceaux, J.; Duddy, W.J.; Duguez, S. Exosomes in Ageing and Motor Neurone Disease: Biogenesis, Uptake Mechanisms, Modifications in Disease and Uses in the Development of Biomarkers and Therapeutics. Cells 2021, 10, 2930. [Google Scholar] [CrossRef] [PubMed]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Lee, Y.; Andaloussi, S.E.; Wood, M.J.A. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, R125–R134. [Google Scholar] [CrossRef]
- Andaloussi, S.E.; Mager, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 85, 177–191. [Google Scholar] [CrossRef]
- Skryabin, G.O.; Komelkov, A.V.; Savelyeva, E.E.; Tchevkina, E.M. Lipid Rafts in Exosome Biogenesis. Biochem. Mosc. 2020, 85, 177–191. [Google Scholar] [CrossRef]
- Donoso-Quezada, J.; Ayala-Mar, S.; González-Valdez, J. The role of lipids in exosome biology and intercellular communication: Function, analytics and applications. Traffic 2021, 22, 204–220. [Google Scholar] [CrossRef]
- Kajimoto, T.; Okada, T.; Miya, S.; Zhang, L.; Nakamura, S. Ongoing activation of sphingosine 1-phosphate receptors mediates maturation of exosomal multivesicular endosomes. Nat. Commun. 2013, 4, 2712. [Google Scholar] [CrossRef] [Green Version]
- Choezom, D.; Gross, J.C. Neutral sphingomyelinase 2 controls exosome secretion by counteracting V-ATPase-mediated endosome acidification. J. Cell Sci. 2022, 135, jcs259324. [Google Scholar] [CrossRef]
- Ayub, M.; Jin, H.K.; Bae, J.S. Novelty of Sphingolipids in the Central Nervous System Physiology and Disease: Focusing on the Sphingolipid Hypothesis of Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 7353. [Google Scholar] [CrossRef] [PubMed]
- Alessenko, A.V.; Albi, E. Exploring Sphingolipid Implications in Neurodegeneration. Front. Neurol. 2020, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, P.; Conte, C.; Albi, E. The Multiple Roles of Sphingomyelin in Parkinson’s Disease. Biomolecules 2021, 11, 1311. [Google Scholar] [CrossRef]
- Podbielska, M.; Ariga, T.; Pokryszko-Dragan, A. Sphingolipid Players in Multiple Sclerosis: Their Influence on the Initiation and Course of the Disease. Int. J. Mol. Sci. 2022, 23, 5330. [Google Scholar] [CrossRef] [PubMed]
- Petit, C.S.; Lee, J.J.; Boland, S.; Swarup, S.; Christiano, R.; Lai, Z.W.; Mejhert, N.; Elliott, S.D.; McFall, D.; Haque, S.; et al. Inhibition of sphingolipid synthesis improves outcomes and survival in GARP mutant wobbler mice, a model of motor neuron degeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 10565. [Google Scholar] [CrossRef]
- Mariosa, D.; Hammar, N.; Malmström, H.; Ingre, C.; Jungner, I.; Ye, W.; Fang, F.; Walldius, G. Blood biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis: A more than 20-year follow-up of the Swedish AMORIS cohort. Ann. Neurol. 2017, 81, 718–728. [Google Scholar] [CrossRef]
- Fernández-Beltrán, L.C.; Godoy-Corchuelo, J.M.; Losa-Fontangordo, M.; Williams, D.; Matias-Guiu, J.; Corrochano, S. A Transcriptomic Meta-Analysis Shows Lipid Metabolism Dysregulation as an Early Pathological Mechanism in the Spinal Cord of SOD1 Mice. Int. J. Mol. Sci. 2021, 22, 9553. [Google Scholar] [CrossRef]
- Bjornevik, K.; Zhang, Z.; O’Reilly, É.; Berry, J.D.; Clish, C.B.; Deik, A.; Jeanfavre, S.; Kato, I.; Kelly, R.S.; Kolonel, L.N.; et al. Prediagnostic plasma metabolomics and the risk of amyotrophic lateral sclerosis. Neurology 2019, 92, e2089–e2100. [Google Scholar] [CrossRef]
- Zhou, J.; Tawk, M.; Tiziano, F.D.; Veillet, J.; Bayes, M.; Nolent, F.; Garcia, V.; Servidei, S.; Bertini, E.; Castro-Giner, F.; et al. Spinal muscular atrophy associated with progressive myoclonic epilepsy is caused by mutations in ASAH1. Am. J. Hum. Genet. 2012, 91, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.O.; Chia, R.; Miller, D.E.; Li, R.; Kumaran, R.; Abramzon, Y.; Alahmady, N.; Renton, A.E.; Topp, S.D.; Gibbs, J.R.; et al. Association of Variants in the SPTLC1 Gene With Juvenile Amyotrophic Lateral Sclerosis. JAMA Neurol. 2021, 78, 1236–1248. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; King, R.; Blake, J.; Groves, M.; Love, S.; Woodward, C.; Hammans, S.; Nicoll, J.; Lennox, G.; O’Donovan, D.G.; et al. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I). Brain 2006, 129 Pt 2, 411–425. [Google Scholar] [CrossRef]
- Kölbel, H.; Kraft, F.; Hentschel, A.; Czech, A.; Gangfuss, A.; Mohassel, P.; Nguyen, C.; Stenzel, W.; Schara-Schmidt, U.; Preuße, C.; et al. New Insights into the Neuromyogenic Spectrum of a Gain of Function Mutation in SPTLC1. Genes 2022, 13, 893. [Google Scholar] [CrossRef] [PubMed]
- Mohassel, P.; Donkervoort, S.; Lone, M.A.; Nalls, M.; Gable, K.; Gupta, S.D.; Foley, A.R.; Hu, Y.; Morales Saute, J.A.; Moreira, A.L.; et al. Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis. Nat. Med. 2021, 27, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Hop, P.J.; Zwamborn, R.A.J.; Hannon, E.; Shireby, G.L.; Nabais, M.F.; Walker, E.M.; Van Rheenen, W.; Van Vugt, J.J.F.A.; Dekker, A.M.; Westeneng, H.J.; et al. Genome-wide study of DNA methylation shows alterations in metabolic, inflammatory, and cholesterol pathways in ALS. Sci. Transl. Med. 2022, 14, eabj0264. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Zhang, S.; Kenna, K.P.; Moll, T.; Franklin, J.P.; Allen, S.; Nezhad, H.G.; Iacoangeli, A.; Yacovzada, N.Y.; Eitan, C.; et al. Rare Variant Burden Analysis within Enhancers Identifies CAV1 as an ALS Risk Gene. Cell Rep. 2020, 33, 108456. [Google Scholar] [CrossRef] [PubMed]
- Šála, M.; Hollinger, K.R.; Thomas, A.G.; Dash, R.P.; Tallon, C.; Veeravalli, V.; Lovell, L.; Kögler, M.; Hřebabecký, H.; Procházková, E.; et al. Novel Human Neutral Sphingomyelinase 2 Inhibitors as Potential Therapeutics for Alzheimer’s Disease. J. Med. Chem. 2020, 63, 6028–6056. [Google Scholar] [CrossRef] [PubMed]
- Anakor, E.; Milla, V.; Connolly, O.; Martinat, C.; Pradat, P.F.; Dumonceaux, J.; Duddy, W.; Duguez, S. The Neurotoxicity of Vesicles Secreted by ALS Patient Myotubes Is Specific to Exosome-Like and Not Larger Subtypes. Cells 2022, 11, 845. [Google Scholar] [CrossRef]
- Ferrara, D.; Pasetto, L.; Bonetto, V.; Basso, M. Role of Extracellular Vesicles in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2018, 12, 574. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, L.; Duddy, W.J.; Martinat, C.; Mariot, V.; Connolly, O.; Milla, V.; Anakor, E.; Ouandaogo, Z.G.; Millecamps, S.; Lainé, J.; et al. Muscle cells of sporadic amyotrophic lateral sclerosis patients secrete neurotoxic vesicles. J. Cachexia Sarcopenia Muscle 2022, 13, 1385–1402. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders—challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T.; Sandhoff, K. Sphingolipid metabolism diseases. Biochim. Biophys. Acta 2006, 1758, 2057–2079. [Google Scholar] [CrossRef]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef] [PubMed]
- Krebs, S.; Medugorac, I.; Röther, S.; Strässer, K.; Förster, M. A missense mutation in the 3-ketodihydrosphingosine reductase FVT1 as candidate causal mutation for bovine spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 6746–6751. [Google Scholar] [CrossRef] [PubMed]
- Borgese, N.; Iacomino, N.; Colombo, S.F.; Navone, F. The Link between VAPB Loss of Function and Amyotrophic Lateral Sclerosis. Cells 2021, 10, 1865. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.M.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet. J. Rare Dis. 2018, 13, 121. [Google Scholar] [CrossRef]
- Regier, D.S.; Proia, R.L.; D’Azzo, A.; Tifft, C.J. The GM1 and GM2 Gangliosidoses: Natural History and Progress toward Therapy. Pediatr. Endocrinol. Rev. 2016, 13 (Suppl. S1), 663–673. [Google Scholar] [PubMed]
- El-Abassi, R.; Singhal, D.; England, J.D. Fabry’s disease. J. Neurol. Sci. 2014, 344, 5–19. [Google Scholar] [CrossRef]
- Biffi, A.; Lucchini, G.; Rovelli, A.; Sessa, M. Metachromatic leukodystrophy: An overview of current and prospective treatments. Bone Marrow Transpl. 2008, 42 (Suppl. S2), S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-Pick diseases. Handb. Clin. Neurol. 2013, 113, 1717–1721. [Google Scholar] [PubMed]
- Barth, B.M.; Shanmugavelandy, S.S.; Tacelosky, D.M.; Kester, M.; Morad, S.A.; Cabot, M.C. Gaucher’s disease and cancer: A sphingolipid perspective. Crit. Rev. Oncog. 2013, 18, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.M.; Bongarzone, E.R.; Sands, M.S. Krabbe disease: New hope for an old disease. Neurosci. Lett. 2021, 752, 135841. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Patin, F.; Descat, A.; Garçon, G.; Corcia, P.; Gelé, P.; Lenglet, T.; Bede, P.; Meininger, V.; Devos, D.; et al. A pharmaco-metabolomics approach in a clinical trial of ALS: Identification of predictive markers of progression. PLoS ONE 2018, 13, e0198116. [Google Scholar] [CrossRef]
- Rozen, S.; Cudkowicz, M.E.; Bogdanov, M.; Matson, W.R.; Kristal, B.S.; Beecher, C.; Harrison, S.; Vouros, P.; Flarakos, J.; Vigneau-Callahan, K.; et al. Metabolomic analysis and signatures in motor neuron disease. Metabolomics 2005, 1, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Querin, G.; Biferi, M.G.; Pradat, P.F. Biomarkers for C9orf7-ALS in Symptomatic and Pre-symptomatic Patients: State-of-the-art in the New Era of Clinical Trials. J. Neuromuscul. Dis. 2022, 9, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Boros, B.D.; Schoch, K.M.; Kreple, C.J.; Miller, T.M. Antisense Oligonucleotides for the Study and Treatment of ALS. Neurotherapeutics 2022. [Google Scholar] [CrossRef] [PubMed]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Bucelli, R.C.; Andrews, J.A.; Otto, M.; Farahany, N.A.; Harrington, E.A.; Chen, W.; Mitchell, A.A.; et al. Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: The ATLAS Study. Neurotherapeutics 2022. [Google Scholar] [CrossRef] [PubMed]
- Lawton, K.A.; Brown, M.V.; Alexander, D.; Li, Z.; Wulff, J.E.; Lawson, R.; Jaffa, M.; Milburn, M.V.; Ryals, J.A.; Bowser, R.; et al. Plasma metabolomic biomarker panel to distinguish patients with amyotrophic lateral sclerosis from disease mimics. Amyotroph Lateral Scler Front. Degener 2014, 15, 362–370. [Google Scholar] [CrossRef]
- Saffari, A.; Cannet, C.; Blaschek, A.; Hahn, A.; Hoffmann, G.F.; Johannsen, J.; Kirsten, R.; Kockaya, M.; Kölker, S.; Müller-Felber, W.; et al. (1)H-NMR-based metabolic profiling identifies non-invasive diagnostic and predictive urinary fingerprints in 5q spinal muscular atrophy. Orphanet. J. Rare Dis. 2021, 16, 441. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Crawford, T.O.; Swoboda, K.J.; Kaufmann, P.; Juhasz, P.; Li, X.; Guo, Y.; Li, R.H.; Trachtenberg, F.; Forrest, S.J.; et al. Candidate proteins, metabolites and transcripts in the Biomarkers for Spinal Muscular Atrophy (BforSMA) clinical study. PLoS ONE 2012, 7, e35462. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Veyrat-Durebex, C.; Bocca, C.; Patin, F.; Vourc’h, P.; Kouassi Nzoughet, J.; Lenaers, G.; Andres, C.R.; Simard, G.; Corcia, P.; et al. Lipidomics Reveals Cerebrospinal-Fluid Signatures of ALS. Sci. Rep. 2017, 7, 17652. [Google Scholar] [CrossRef] [PubMed]
- Lawton, K.A.; Cudkowicz, M.E.; Brown, M.V.; Alexander, D.; Caffrey, R.; Wulff, J.E.; Bowser, R.; Lawson, R.; Jaffa, M.; Milburn, M.V.; et al. Biochemical alterations associated with ALS. Amyotroph. Lateral Scler. 2012, 13, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Boss, J.; Guo, K.; Alakwaa, F.M.; Patterson, A.; Kim, S.; Savelieff, M.G.; Hur, J.; Feldman, E.L. Untargeted metabolomics yields insight into ALS disease mechanisms. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Guo, K.; Savelieff, M.G.; Patterson, A.; Sakowski, S.A.; Habra, H.; Karnovsky, A.; Hur, J.; Feldman, E.L. Metabolomics identifies shared lipid pathways in independent amyotrophic lateral sclerosis cohorts. Brain 2022, awac025. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Lin, C.N.; Chen, C.M.; Lyu, R.K.; Chu, C.C.; Liao, M.F.; Huang, C.C.; Chang, H.S.; Ro, L.S.; Kuo, H.C. Altered Metabolic Profiles of the Plasma of Patients with Amyotrophic Lateral Sclerosis. Biomedicines 2021, 9, 1944. [Google Scholar] [CrossRef] [PubMed]
- FernÁndez-Eulate, G.; Ruiz-Sanz, J.I.; Riancho, J.; ZufirÍa, M.; GereÑu, G.; FernÁndez-TorrÓn, R.; Poza-Aldea, J.J.; Ondaro, J.; Espinal, J.B.; GonzÁlez-ChinchÓn, G.; et al. A comprehensive serum lipidome profiling of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. Front. Degener 2020, 21, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.C.; Treleaven, C.M.; Pacheco, J.; Cooper, S.; Bao, C.; Abraham, M.; Cromwell, M.; Sardi, S.P.; Chuang, W.L.; Sidman, R.L.; et al. Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2015, 112, 8100–8105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sol, J.; Jové, M.; Povedano, M.; Sproviero, W.; Domínguez, R.; Piñol-Ripoll, G.; Romero-Guevara, R.; Hye, A.; Al-Chalabi, A.; Torres, P.; et al. Lipidomic traits of plasma and cerebrospinal fluid in amyotrophic lateral sclerosis correlate with disease progression. Brain Commun. 2021, 3, fcab143. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Larrea, D.; Yun, T.; Xu, Y.; Hupf, J.; Zandkarimi, F.; Chan, R.B.; Mitsumoto, H. Lipidomics study of plasma from patients suggest that ALS and PLS are part of a continuum of motor neuron disorders. Sci. Rep. 2021, 11, 13562. [Google Scholar] [CrossRef]
- Bouscary, A.; Quessada, C.; René, F.; Spedding, M.; Turner, B.J.; Henriques, A.; Ngo, S.T.; Loeffler, J.P. Sphingolipids metabolism alteration in the central nervous system: Amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases. Semin. Cell Dev. Biol. 2021, 112, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Pournajaf, S.; Dargahi, L.; Javan, M.; Pourgholami, M.H. Molecular Pharmacology and Novel Potential Therapeutic Applications of Fingolimod. Front. Pharmacol. 2022, 13, 807639. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.F.; Bowen, J.D.; Reder, A.T. The Direct Effects of Fingolimod in the Central Nervous System: Implications for Relapsing Multiple Sclerosis. CNS Drugs 2016, 30, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Groves, A.; Kihara, Y.; Chun, J. Fingolimod: Direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J. Neurol. Sci. 2013, 328, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.D.; Paganoni, S.; Atassi, N.; Macklin, E.A.; Goyal, N.; Rivner, M.; Simpson, E.; Appel, S.; Grasso, D.L.; Mejia, N.I.; et al. Phase IIa trial of fingolimod for amyotrophic lateral sclerosis demonstrates acceptable acute safety and tolerability. Muscle Nerve 2017, 56, 1077–1084. [Google Scholar] [CrossRef]
- Zhu, C.; Bilousova, T.; Focht, S.; Jun, M.; Elias, C.J.; Melnik, M.; Chandra, S.; Campagna, J.; Cohn, W.; Hatami, A.; et al. Pharmacological inhibition of nSMase2 reduces brain exosome release and α-synuclein pathology in a Parkinson’s disease model. Mol. Brain 2021, 14, 70–79. [Google Scholar] [CrossRef]
- Bouscary, A.; Quessada, C.; Mosbach, A.; Callizot, N.; Spedding, M.; Loeffler, J.P.; Henriques, A. Ambroxol Hydrochloride Improves Motor Functions and Extends Survival in a Mouse Model of Familial Amyotrophic Lateral Sclerosis. Front. Pharmacol. 2019, 10, 883. [Google Scholar] [CrossRef]
- Laurila, P.P.; Luan, P.; Wohlwend, M.; Zanou, N.; Crisol, B.; Imamura de Lima, T.; Goeminne, L.J.E.; Gallart-Ayala, H.; Shong, M.; Ivanisevic, J.; et al. Inhibition of sphingolipid de novo synthesis counteracts muscular dystrophy. Sci. Adv. 2022, 8, eabh4423. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Gene | Affected Enzyme/Protein | Effect on Sphingolipids |
|---|---|---|---|
| Sphingolipid synthesis | |||
| Juvenile ALS [92] HSAN1 [93] | SPTLC1 | SPT | Atypical deoxysphingolipids, cannot be converted into complex SLs or degraded |
| Bovine SMA [106] | FVT1 | KSR | Reduced ceramide synthesis from de novo pathway |
| ALS type 8 [107] Late onset SMA [108] | VAPB | VAPB with effect on CERT and FAPP2 | Impaired transfer of ceramide and glucosylceramide from ER to golgi apparatus |
| ALS [96] | SGMS2 | SMS2 | Affects sphingomyelin synthesis |
| Sphingolipid degradation | |||
| SMA-PME [91] Farber’s disease [109] | ASAH1 | Acid ceramidase | Ceramide accumulation |
| GM1 gangliodosis [110] | GLB1 | β-Galactosidase | GM1 ganglioside accumulation |
GM2 gangliodoses [110]
| HEXA HEXB |
| GM2 ganglioside accumulation GM2 ganglioside, glycolipid GA2 and globoside accumulation |
| Fabry’s Disease [111] | GLA | α-Galactosidase A | Globotriaosylceramide accumulation |
| Metachromatic Leukodystrophy [112] | ARSA | Arylsulphatase A | Sulfatides accumulation |
Niemann-Pick Disease [113]
| SMPD1 NPC1/NPC2 | Sphingomyelinase | Sphingomyelin accumulation |
| Gaucher’s Disease [114] | GBA | Glucocerebrosidase | Glucosylceramide accumulation |
| Krabbe’s Disease [115] | GALC | Galactosylceramidase | Galactosylceramide accumulation |
| Study | Patients | Sample Type | Quantification Platform | Metabolites Evaluated | Lipid Changes in MND | Prognostic Use |
|---|---|---|---|---|---|---|
| Blasco et al. 2017 [125] | 40 ALS 45 Controls | CSF | HRMS | 122 lipids | ↑: PC (36:4p), PC (36:4e), SM (d43:2), SM (d34:0) | Higher SM (d43:2) and lower TG (16:0/16:0/18:1) and TG (18:0/16:0/18:1) had slower progression |
| ↓: TG (16:1/18:1/18:2) | ||||||
| Lawton et al. 2012 [126] | 161 ALS 117 Controls | Plasma | GC/MS and UPLC-MS/MS | 335 lipids, proteins and carbohydrates | ↑: LPC (16:1) and SM (18:0) | Not evaluated |
| Cutler et al. 2002 [16] | 9 ALS 3 Control | Spinal cord | ES/MS/MS | Sphingolipids, Phospholipids, Cholesterol Esters, and Lipid Peroxides | ↑: Cer (C16:0), Cer (C24:0), SM (C16:0), CE (C16:0) and CE (C18:0) | Not evaluated |
| Goutman et al. 2020 [127] | 125 ALS 71 Controls | Plasma | UPLC-MS/MS | 899 metabolites | ↑: 8 Cers, 28 DAGs, 5 HEXC, 24 SMs, | Not evaluated |
| ↓: 5 DAGs, 5 SMs | ||||||
| Goutman et al. 2022 [128] | Above cohort of 125 ALS and 71 controls with 2nd cohort 225 ALS, 104 controls | Plasma | UPLC-MS/MS | 640 metabolites | SM most significant sub-pathway LCFA, acyl intermediates and Cers also raised | SM (d18:1/24:0), SM (d18:1/20:0, d16:1/22:0), SM (d18:1/14:0, d16:1/16:0) and lignoceroylcarnitine (C24) correlated with ALSFRS-R |
| Bjornevik et al. 2019 [90] | 275 ALS 549 Controls | Plasma | LC/MS | 404 metabolites | ↑: SM (C18:2), PC (C40:7), PC (C38:4), CE (C22:4) | Not evaluated |
| ↓: 12 TAGs, DAG (C36:1), DAG (C36:2), PC (C36:2), 21-deoxycortisol, butyrobetaine | ||||||
| Lawton et al. 2014 [122] | 172 ALS 73 neurological mimics 50 Controls | plasma | GC/MS and UPLC-MS/MS | 367 metabolites | ↑: SM (d18:1/16:0), 5 FAs, 3-dehydrocarnitine, 1,2-propanediol, Chol, 1-stearoyl-GPI | 1,2-propanediol correlated with ALSFRS-R |
| Chang et al. 2021 [129] | 36 ALS 36 Controls | plasma | LC–MS/MS | 185 metabolites | ↑: SM (C24:1), SM (C20:2), PC (C44:5), PC (C34:2) | 14 PCs and (OH) SM(C22:1) correlated with ALSFRS-R |
| ↓: (OH) SM(C22:1) (OH) SM(C24:1) 29 other PCs | ||||||
| Fernandez-Eulate et al. 2020 [130] | 20 ALS 20 Controls | Serum | UPLC-MS | 416 lipids | ↑: SM (39:1), SM (33:1), PE (P-20:1/0:0), PE (O-16:0/0:0), 5 PCs, androsterone, etiocholanolone and 2 FAs | Not evaluated |
| Blasco et al. 2018 [116] | 74 ALS | Plasma | HPLC-MS/MS | 188 metabolites | Not evaluated—no control participants | SM (C22:3) and SM (C34:1) correlated with disease progression, SM (24:1), SM (C16:1) and (OH) SM (C22:2) correlated with SVC |
| Dodge et al. 2015 [131] | 6 ALS 6 Control | Spinal cord | LC-MS/MS | Cer, SM and GSLs | ↑: Cer (C18:0), Cer (C24:1), (OH) Cer (C24:0), Cerebroside (C18:0 and C24:1), GlcCer (C18:0 and C24:1), LacCer (18:0), GL3 (C22:1), GM3 (C23:0), GM1 (C18:0) AND SM (C18:0) | Not evaluated |
| Sol et al. 2021 [132] | 23 ALS 10 Controls | CSF Plasma | LC-MS/MS | 1018 lipids in plasma and 843 in CSF | ↑: 3 Fas, 2 DAGs, 13 TGs, 17 GPLs, 3 Cer, 1 SM | Fast vs. slow progressors had increased- 1 FA, 4 GLs, 4 GPLs, 2 Cer, 1 GM3, and decreased- 46 GLs, 36 GPLs, 2 Cer, 8 SM, 5 CE |
| ↓: 2 DAGs, 4 GPLs, 3 Cer, 3 GLs | ||||||
| Area-Gomez et al. 2021 [133] | 40 ALS 28 PLS 28 Control | Serum/Plasma | LC/MS | 532 lipids | ↑: Cer, LacCer, CE | SM declined and Cer increased at follow up |
| ↓: SM, PC, PS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCluskey, G.; Donaghy, C.; Morrison, K.E.; McConville, J.; Duddy, W.; Duguez, S. The Role of Sphingomyelin and Ceramide in Motor Neuron Diseases. J. Pers. Med. 2022, 12, 1418. https://doi.org/10.3390/jpm12091418
McCluskey G, Donaghy C, Morrison KE, McConville J, Duddy W, Duguez S. The Role of Sphingomyelin and Ceramide in Motor Neuron Diseases. Journal of Personalized Medicine. 2022; 12(9):1418. https://doi.org/10.3390/jpm12091418
Chicago/Turabian StyleMcCluskey, Gavin, Colette Donaghy, Karen E. Morrison, John McConville, William Duddy, and Stephanie Duguez. 2022. "The Role of Sphingomyelin and Ceramide in Motor Neuron Diseases" Journal of Personalized Medicine 12, no. 9: 1418. https://doi.org/10.3390/jpm12091418