
Primary Synovial Sarcoma of the Kidney: Diagnostic Approach and Therapeutic Modalities for a Rare Nosological Entity
 , , , , ,
, , , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
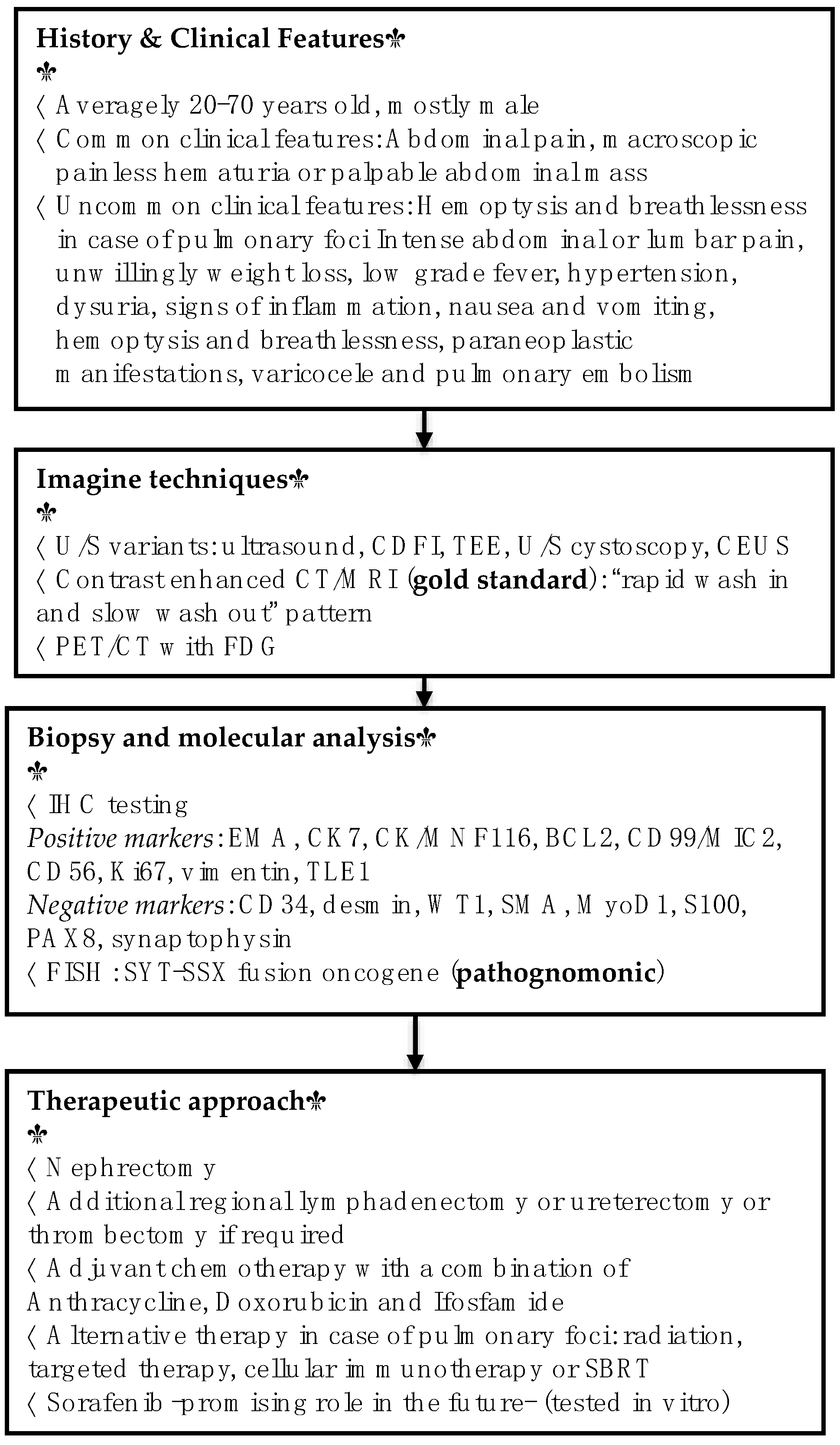
2. Epidemiology and Classification
3. Clinical Features
4. Histopathology and Immunochemistry
5. Diagnostic Modalities
6. Therapeutic Approach
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cai, H.J.; Cao, N.; Wang, W.; Kong, F.L.; Sun, X.X.; Huang, B. Primary renal synovial sarcoma: A case report. World J. Clin. Cases 2019, 7, 3098–3103. [Google Scholar] [CrossRef] [PubMed]
- Pitino, A.; Squillaci, S.; Spairani, C.; Cosimi, M.F.; Feyles, E.; Ricci, D.; Bardari, F.; Graziano, M.; Morabito, F.; Cesarani, F.; et al. Primary synovial sarcoma of the kidney. A case report with pathologic appraisal investigation and literature review. Pathologica 2011, 103, 271–278. [Google Scholar] [PubMed]
- Romero-Rojas, A.E.; Díaz-Pérez, J.A.; Messa-Botero, O.A.; Neira-Mejia, F.E. Early age renal synovial sarcoma. Arch. Esp. Urol. 2010, 63, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, S.; Tsuru, T.; Okamoto, K.; Narita, M.; Okada, Y. Primary synovial sarcoma arising from a crossed ectopic kidney with fusion. Int. J. Urol. 2010, 17, 96–98. [Google Scholar] [CrossRef]
- Wang, Z.H.; Wang, X.C.; Xue, M. Clinicopathologic analysis of 4 cases of primary renal synovial sarcoma. Chin. J. Cancer 2010, 29, 212–216. [Google Scholar] [CrossRef]
- Gulum, M.; Yeni, E.; Savas, M.; Ozardali, I.; Ozdemir, I.; Mil, D.; Altunkol, A.; Ciftci, H. Primary renal synovial sarcoma. Case Rep. Urol. 2011, 2011. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sohn, J.H.; Lee, M.C.; Lee, G.; Yoon, G.-S.; Hashimoto, H.; Sonobe, H.; Ro, J.Y. Primary synovial sarcoma of the kidney. Am. J. Surg. Pathol. 2000, 24, 1097–1104. [Google Scholar] [CrossRef]
- Ozkan, E.E.; Mertsoylu, H.; Ozardali, H.I. A case of renal synovial sarcoma treated with adjuvant ifosfamide and doxorubicin. Intern. Med. 2011, 50, 1575–1580. [Google Scholar] [CrossRef]
- Chen, S.; Bhuiya, T.; Liatsikos, E.N.; Alexianu, M.D.; Weiss, G.H.; Kahn, L.B. Primary synovial sarcoma of the kidney: A case report with literature review. Int. J. Surg. Pathol. 2011, 9, 335–339. [Google Scholar] [CrossRef]
- Xu, R.F.; He, E.H.; Yi, Z.X.; Lin, J.; Zhang, Y.N.; Qian, L.X. Multimodality-imaging manifestations of primary renal-allograft synovial sarcoma: First case report and literature review. World J. Clin. Cases 2019, 27, 1677–1685. [Google Scholar] [CrossRef]
- Divetia, M.; Karpate, A.; Basak, R.; Desai, S.B. Synovial sarcoma of the kidney. Ann. Diagn. Pathol. 2008, 12, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhong, Z.; Zhu, L.; Xiong, W.; Pan, C.; Wang, X.; Huang, Z.; Zhao, X. Primary synovial sarcoma of the kidney: A case report. Oncol. Lett. 2015, 10, 3542–3544. [Google Scholar] [CrossRef] [PubMed]
- Scarpato, K.R.; Makari, J.H.; Agaronov, M.; Balarezo, F.; Parikh, N.; Finck, C.M.; Ferrer, F.A. Primary renal synovial sarcoma in a 13-year-old boy. J. Pediatric Surg. 2011, 46, 1849–1851. [Google Scholar] [CrossRef] [PubMed]
- Dutt, U.K.; Manikandan, R.; Dorairajan, L.N.; Srinivas, B.H. Biphasic renal synovial sarcoma with extensive venous tumor thrombosis: A rare presentation. Urol. Ann. 2018, 10, 339–341. [Google Scholar]
- Huang, Y.; Liu, D.; Luo, J.; Chen, W. Primary renal synovial sarcoma: A case report and literature review. J. Cancer Res. Ther. 2018, 14, 267–269. [Google Scholar]
- Vedana, M.; Fuenfschilling, M.; Tzankov, A.; Zellweger, T. Primary synovial cell sarcoma of the kidney: Case report and review of the literature. Case Rep. Oncol. 2015, 8, 128–132. [Google Scholar] [CrossRef]
- Yang, L.; Wang, K.; Hong, L.; Wang, Y.; Li, X. The value of immunohistochemistry in diagnosing primary renal synovial sarcoma: A case report and literature review. Int. Sur. 2012, 97, 177–181. [Google Scholar] [CrossRef]
- Nishida, T.; Inamoto, T.; Uehara, H.; Ibuki, N.; Koyama, K.; Komura, K.; Fujisue, Y.; Kurisu, Y.; Tsuji, M.; Azuma, H.; et al. Monophasic primary renal synovial sarcoma accompanied with a hemorrhagic cyst. Urol. J. 2011, 8, 244–247. [Google Scholar]
- Wang, Y.; Wang, K.; Zhang, Z.; Chen, L.; Xu, F. Primary renal synovial sarcoma-a case report. West Indian Med. J. 2011, 60, 354–356. [Google Scholar]
- El Chediak, A.; Mukherji, D.; Temraz, S.; Temraz, S.; Nassif, S.; Sinno, S.; Mahfouz, R.; Shamseddine, A. Primary synovial sarcoma of the kidney: A case report of complete pathological response at a Lebanese tertiary care center. BMC Urol. 2018, 18. [Google Scholar] [CrossRef]
- Modi, G.; Madabhavi, I.; Panchal, H.; Anand, A.; Patel, A.; Parikh, S.; Revannasiddaiah, S. Primary synovial sarcoma of kidney: A rare differential diagnosis of renomegaly. Case Rep. Pathol. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Rose, L.; Grignon, D.; Cheng, L.; Fan, R.; Zhang, S.; Alruwaii, F.; Chen, S. Primary Renal Synovial Sarcomas: PAX 8 Immunostaining and Unusual Molecular Findings. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, D.; Narayanaswamy, K.; Sundersingh, S.; Senniappan, K.; Raja, A. Primary Synovial Sarcoma of the Kidney with Inferior Vena Caval Thrombus. Indian J. Surg. Oncol. 2016, 7, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Kang, W.; Li, S.; Yang, Z.; Xu, J. CT findings of synovial sarcomas of the kidney with pathological correlation. Clin. Imaging 2013, 37, 1033–1036. [Google Scholar] [CrossRef]
- Blas, L.; Roberti, J. Primary Renal Synovial Sarcoma and Clinical and Pathological Findings: A Systematic Review. Curr. Urol. Rep. 2021, 22, 25. [Google Scholar] [CrossRef]
- Abbas, M.; Dämmrich, M.E.; Braubach, P.; Meinardus, A.; Kramer, M.W.; Merseburger, A.S.; Herrmann, T.R.W.; Grünwald, V.; Kreipe, H.-H. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare Tumors 2014, 6, 5393. [Google Scholar] [CrossRef]
- Marković-Lipkovski, J.; Sopta, J.; Vjestica, J.; Vujanić, G.; Tulić, C. Rapidly progressive course of primary renal synovial sarcoma-case report. Srp. Arh. Za Celok. Lek. 2013, 141, 814–818. [Google Scholar] [CrossRef]
- Dassi, V.; Das, K.; Singh, B.P.; Swain, S.K. Primary synovial sarcoma of kidney: A rare tumor with an atypical presentation. Indian J. Urol. 2009, 25, 269–271. [Google Scholar] [CrossRef]
- Iacovelli, R.; Altavilla, A.; Ciardi, A.; Urbano, F.; Manai, C.; Gentile, V.; Cortesi, E. Clinical and pathological features of primary renal synovial sarcoma: Analysis of 64 cases from 11 years of medical literature. BJU Int. 2012, 110, 1449–1454. [Google Scholar] [CrossRef]
- Pathrose, G.; John, N.T.; Hariharan, P. Renal Synovial Sarcoma in a Young Pregnant Lady: A Case Report and Clinico-Pathological Profile. J Clin. Diagn. Res. 2017, 11. [Google Scholar] [CrossRef]
- Bakhshi, G.D.; Khan, A.S.; Shaikh, A.S.; Khan, M.A.; Khan, M.A.; Jamadar, N.M. Primary renal synovial sarcoma. Clins Pract. 2012, 2, e44. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, H. Prognostic features of renal sarcomas (Review). Oncol. Lett. 2015, 9, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Vesoulis, Z.; Rahmeh, T.; Nelson, R.; Clarke, R.; Lu, Y.; Dankoff, J. Fine needle aspiration biopsy of primary renal synovial sarcoma. A case report. Acta. Cytol. 2003, 47, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, A.E.; Saunders, S.E.; Zaslau, S.; Chang, W.W.; Farivar-Mohseni, H. Primary synovial sarcoma of the kidney. Int. J. Urol. 2005, 12, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.F.; Qiu, Y.W.; Han, L.J.; Cao, J.; Zhang, C.; Liu, Z.-Y.; Zhang, X.-L.; Cai, P.-Q.; Li, L. Primary renal synovial sarcoma: Computed tomography imaging findings. Acta Radiol. 2015, 56, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Wezel, F.; Ströbel, P.; Michaely, H.; Michel, M.S.; Häcker, A. Biphasisches primäres Synovialsarkom der Niere. Der Urol. 2010, 49, 411–414. [Google Scholar] [CrossRef]
- Dewana, S.K.; Parmar, K.M.; Sharma, G.; Bansal, A.; Panwar, P.; Mavuduru, R.S. Paraneoplastic hepatic dysfunction with jaundice in a case of primary renal synovial sarcoma: A very rare scenario. Urol. Case Rep. 2019, 24. [Google Scholar] [CrossRef]
- Lakshmaiah, K.C.; Saini, K.S.; Singh, T.; Jain, A.; Kumar, R.V.; Sajeevan, K.V.; Lokanatha, D.; Jacob, L.A. Primary synovial sarcoma of kidney-a report of 2 cases and review of literature. J. Egypt. Natl. Cancer Inst. 2010, 22, 149–153. [Google Scholar]
- Radhakrishnan, V.; Dhanushkodi, M.; Narayanswamy, K.; Raja, A.; Sundersingh, S.; Sagar, T. Synovial sarcoma of kidney in a child: A rare presentation. J. Indian Assoc. Pediatr. Surg. 2016, 21, 75–77. [Google Scholar] [CrossRef]
- Kataria, T.; Janardhan, N.; Abhishek, A.; Sharan, G.K.; Mitra, S. Pulmonary metastasis from renal synovial sarcoma treated by stereotactic body radiotherapy: A case report and review of the literature. J. Cancer Res. Ther. 2010, 6, 75–79. [Google Scholar] [CrossRef]
- Trolliet, S.; Lindner, V.; Krzisch, S.; Schneider, M.; Jung, J.L. Synovialosarcome rénal primitive. Prog. Urol. 2014, 24, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Puj, K.S.; Pandya, S.J.; Warikoo, V.; Chauhan, T.; Samanta, S.T. Primary Synovial Sarcoma of the Kidney. A Rare Presentat. Urol. 2018, 116. [Google Scholar]
- Lopes, H.; Pereira, C.A.; Zucca, L.E.; Serrano, S.V.; Silva, S.R.M.; Camparoto, M.L.; Cárcano, F.M. Primary monophasic synovial sarcoma of the kidney: A case report and review of literature. Clin. Med. Insights Oncol. 2013, 7, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Dey, S.; Khandakar, B.; Medda, S.; Chandra Paul, P. Primary renal synovial sarcoma: A rare tumor with an atypical presentation. Arch. Iran. Med. 2014, 17, 726–728. [Google Scholar] [PubMed]
- Mishra, S.; Awasthi, N.; Hazra, S.P.; Bera, M.K. Primary synovial sarcoma of the kidney. Saudi J. Kidney Dis. Transpl. 2015, 26, 996–999. [Google Scholar] [CrossRef]
- Schoolmeester, J.K.; Cheville, J.C.; Folpe, A.L. Synovial sarcoma of the kidney: A clinicopathologic, immunohistochemical, and molecular genetic study of 16 cases. Am. J. Surg. Pathol. 2014, 38, 60–65. [Google Scholar] [CrossRef]
- Ozkanli, S.S.; Yildirim, A.; Zemheri, E.; Gucer, F.I.; Aydin, A.; Caskurlu, T. Primary synovial sarcoma of the kidney. Urol. Int. 2012, 92, 369–372. [Google Scholar] [CrossRef]
- Karafin, M.; Parwani, A.V.; Netto, G.J.; Illei, P.B.; Epstein, J.I.; Ladanyi, M.; Argani, P. Diffuse expression of PAX2 and PAX8 in the cystic epithelium of mixed epithelial stromal tumor, angiomyolipoma with epithelial cysts, and primary renal synovial sarcoma: Evidence supporting renal tubular differentiation. Am. J. Surg. Pathol. 2011, 35, 1264–1273. [Google Scholar] [CrossRef]
- Rekhi, B.; Basak, R.; Desai, S.B.; Jambhekar, N.A. Immunohistochemical validation of TLE1, a novel marker, for synovial sarcomas. Indian J. Med. Res. 2012, 136, 766–775. [Google Scholar]
- Bella, A.J.; Winquist, E.W.; Perlman, E.J. Primary synovial sarcoma of the kidney diagnosed by molecular detection of SYT-SSX fusion transcripts. J. Urol. 2002, 168, 1092–1093. [Google Scholar] [CrossRef]
- Koyama, S.; Morimitsu, Y.; Morokuma, F.; Hashimoto, H. Primary synovial sarcoma of the kidney: Report of a case confirmed by molecular detection of the SYT-SSX2 fusion transcripts. Pathol. Int. 2001, 51, 385–391. [Google Scholar] [CrossRef]
- Chen, P.C.; Chang, Y.H.; Yen, C.C.; Pan, C.C.; Chiang, H. Primary renal synovial sarcoma with inferior vena cava and right atrium invasion. Int. J. Urol. 2003, 10, 657–660. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.; Sekiguchi, Z.; Makiyama, K.; Nakayama, T.; Nagashima, Y.; Kita, K.; Namura, K.; Itou, H.; Sano, F.; Hayashi, N.; et al. Primary Synovial Sarcoma of the Kidney. Case Rep. Oncol. 2009, 2, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Zakhary, M.M.; Elsayes, K.M.; Platt, J.F.; Francis, I.R. Magnetic resonance imaging features of renal synovial sarcoma: A case report. Cancer Imaging 2008, 8, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Erturhan, S.; Seçkiner, I.; Zincirkeser, S.; Erbaǧci, A.; Çelik, M.; Yaǧci, F.; Karakok, M. Primary synovial sarcoma of the kidney: Use of PET/CT in diagnosis and follow-up. Ann. Nucl. Med. 2008, 22, 225–229. [Google Scholar] [CrossRef]
- Pereira, E.; Silva, R.; Leitão, T.; Correia, L.; Martins, F.; Palma Dos Reis, J.; Lopes, T. Primary synovial sarcoma of the kidney with unusual follow up findings. Can. J. Urol. 2013, 20, 6734–6736. [Google Scholar]
- Tan, Y.S.; Ng, L.G.; Yip, S.K.; Tay, M.H.; Lim, A.S.; Tien, S.L.; Cheng, L.; Tan, P.H. Synovial sarcoma of the kidney: A report of 4 cases with pathologic appraisal and differential diagnostic review. Anal. Quant. Cytol. Histol. 2010, 32, 239–245. [Google Scholar]
- Park, S.J.; Kim, H.K.; Kim, C.K.; Park, S.-K.; Go, E.-S.; Kim, M.-E.; Hong, D.S. A case of renal synovial sarcoma: Complete remission was induced by chemotherapy with doxorubicin and ifosfamide. Korean J. Intern. Med. 2004, 19, 62–65. [Google Scholar] [CrossRef]
- Mirza, M.; Zamilpa, I.; Bunning, J. Primary renal synovial sarcoma. Urology 2008, 72, 716.e11–716.e12. [Google Scholar] [CrossRef] [PubMed]
- Törnkvist, M.; Wejde, J.; Ahlén, J.; Brodin, B.; Larsson, O. A novel case of synovial sarcoma of the kidney: Impact of SS18/SSX analysis of renal hemangiopericytoma-like tumors. Diagn. Mol. Pathol. 2004, 13, 47–51. [Google Scholar] [CrossRef]
- Paláu, L.M.A.; Thu Pham, T.; Barnard, N.; Merino, M.J. Primary synovial sarcoma of the kidney with rhabdoid features. Int. J. Surg. Pathol. 2007, 15, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Shannon, B.A.; Murch, A.; Cohen, R.J. Primary renal synovial sarcoma confirmed by cytogenetic analysis: A lesion distinct from sarcomatoid renal cell carcinoma. Arch. Pathol. Lab. Med. 2005, 12, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Faria, P.A.; Epstein, J.I.; Reuter, V.E.; Perlman, E.J.; Beckwith, J.B.; Ladanyi, M. Primary renal synovial sarcoma: Molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am. J. Surg. Pathol. 2000, 24, 1087–1096. [Google Scholar] [CrossRef]
- Grampurohit, V.U.; Myageri, A.; Rao, R.V. Primary renal synovial sarcoma. Urol. Ann. 2011, 3, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.D.; Huang, K.H.; Chueh, S.C.; Lai, M.K.; Lin, W.C. Primary synovial sarcoma of the kidney. J. Med. Assoc. 2008, 107, 344–347. [Google Scholar] [CrossRef]
- Houdta, W.J.; Rautb, C.P.; Bonvalotc, S.; Swallowd, C.J.; Haase, R.; Gronchi, A. New research strategies in retroperitoneal sarcoma. The case of TARPSWG, STRASS and RESAR: Making progress through collaboration. Curr. Opin. Oncol. 2019, 31, 310–316. [Google Scholar] [CrossRef]
- Chi, Y.; Fang, Z.; Hong, X.; Yao, Y.; Sun, P.; Wang, G.; Du, F.; Sun, Y.; Wu, Q.; Qu, G.; et al. Safety and Efficacy of Anlotinib, a Multikinase Angiogenesis Inhibitor, in Patients with Refractory Metastatic Soft-Tissue Sarcoma. Clin. Cancer Res. 2018, 24, 5233–5238. [Google Scholar] [CrossRef]
- Machairas, N.; Tsilimigras, D.I.; Pawlik, T.M. Current Landscape of Immune Checkpoint Inhibitor Therapy for Hepatocellular Carcinoma. Cancers 2022, 14, 2018. [Google Scholar] [CrossRef]
- Mosca, L.; de Angelis, A.; Ronchi, A.; de Chiara, A.; Fazioli, F.; Ruosi, C.; Altucci, L.; Conte, M.; de Nigris, F. Sarcoma Common MHC-I Haplotype Restricts Tumor-Specific CD8+ T Cell Response. Cancers 2022, 14, 3414. [Google Scholar] [CrossRef]
- Basso, U.; Brunello, A.; Bertuzzi, A.; Santoro, A. Sorafenib is active on lung metastases from synovial sarcoma. Ann. Oncol. 2009, 20, 386–387. [Google Scholar] [CrossRef]
- Peng, C.L.; Guo, W.; Ji, T.; Ren, T.; Yang, Y.; Li, D.-S.; Qu, H.y.; Li, X.; Tang, S.; Yan, T.-Q.; et al. Sorafenib induces growth inhibition and apoptosis in human synovial sarcoma cells via inhibiting the RAF/MEK/ERK signaling pathway. Cancer Biol. Ther. 2009, 8, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Mastoraki, A.; Schizas, D.; Papanikolaou, I.S.; Bagias, G.; Machairas, N.; Agrogiannis, G.; Liakakos, T.; Liakakos, T.; Arkadopoulos, N. Management of primary retroperitoneal synovial sarcoma: A case report and review of literature. World. J. Gastrointest. Surg. 2019, 11, 27–33. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mastoraki, A.; Schizas, D.; Karavolia, D.M.; Smailis, A.; Machairas, N.; Vailas, M.; Michalinos, A.; Tsapralis, D.; Anastasiou, I.; Agrogiannis, G. Primary Synovial Sarcoma of the Kidney: Diagnostic Approach and Therapeutic Modalities for a Rare Nosological Entity. J. Pers. Med. 2022, 12, 1450. https://doi.org/10.3390/jpm12091450
Mastoraki A, Schizas D, Karavolia DM, Smailis A, Machairas N, Vailas M, Michalinos A, Tsapralis D, Anastasiou I, Agrogiannis G. Primary Synovial Sarcoma of the Kidney: Diagnostic Approach and Therapeutic Modalities for a Rare Nosological Entity. Journal of Personalized Medicine. 2022; 12(9):1450. https://doi.org/10.3390/jpm12091450
Chicago/Turabian StyleMastoraki, Aikaterini, Dimitrios Schizas, Despoina Maria Karavolia, Antonios Smailis, Nikolaos Machairas, Michail Vailas, Adamantios Michalinos, Dimitrios Tsapralis, Ioannis Anastasiou, and George Agrogiannis. 2022. "Primary Synovial Sarcoma of the Kidney: Diagnostic Approach and Therapeutic Modalities for a Rare Nosological Entity" Journal of Personalized Medicine 12, no. 9: 1450. https://doi.org/10.3390/jpm12091450