
Plasma Angiotensin Converting Enzyme 2 (ACE2) Activity in Healthy Controls and Patients with Cardiovascular Risk Factors and/or Disease
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
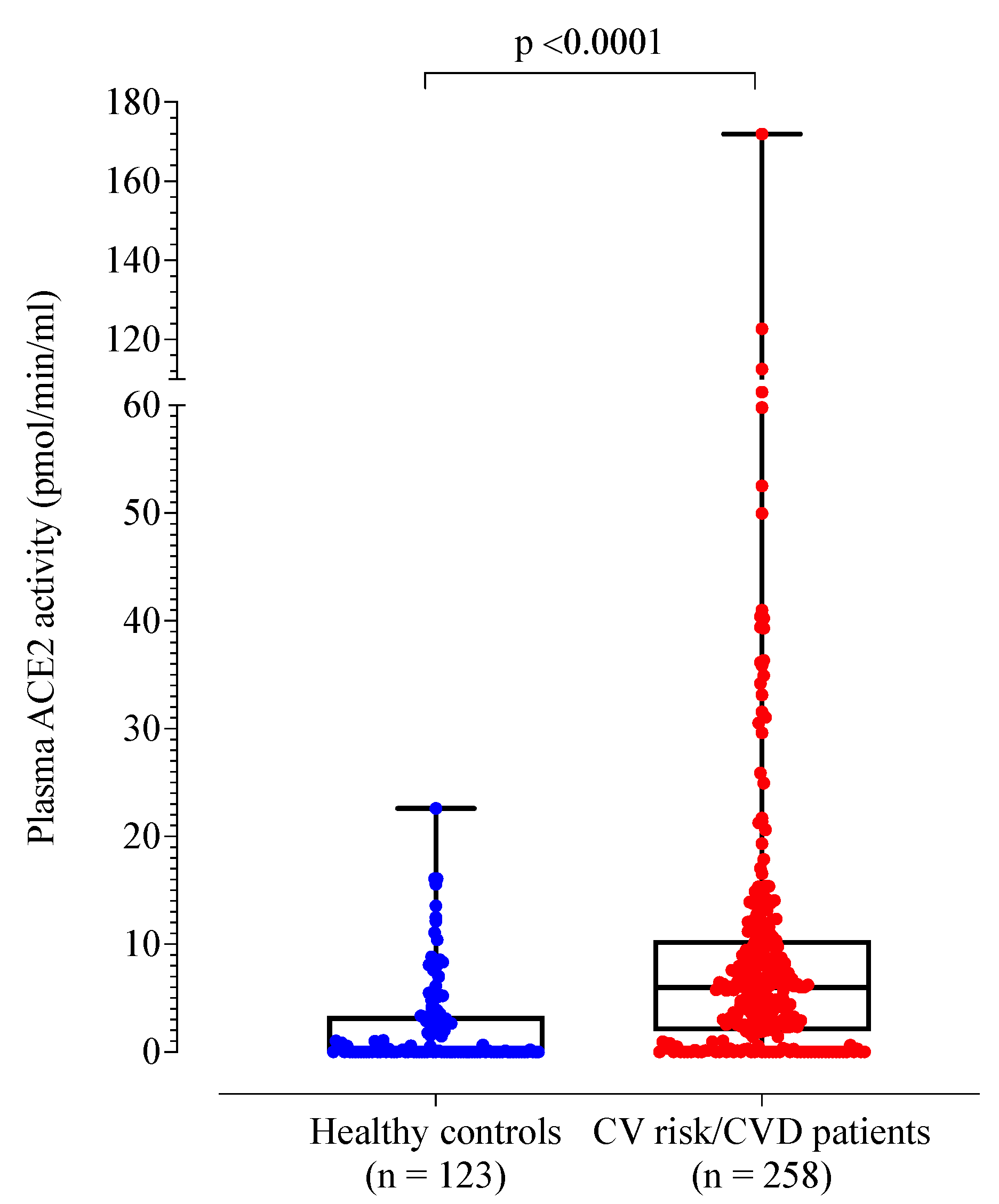
3. Results
3.1. Healthy Controls
3.2. Patients with Cardiovascular Risk Factors and/or Disease Compared to Healthy Controls
3.3. Analysis of Patients with Cardiovascular Risk Factors and/or Disease according to Tertiles
4. Discussions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lew, R.A.; Warner, F.J.; Hanchapola, I.; Yarski, M.A.; Ramchand, J.; Burrell, L.M.; Smith, A.I. Angiotensin-converting enzyme 2 catalytic activity in human plasma is masked by an endogenous inhibitor. Exp. Physiol. 2008, 93, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Crackower, M.A.; Backx, P.H.; Penninger, J.M. The role of ACE2 in cardiovascular physiology. Trends Cardiovasc. Med. 2003, 13, 93–101. [Google Scholar] [CrossRef]
- Hamming, I.; Cooper, M.E.; Haagmans, B.L.; Hooper, N.M.; Korstanje, R.; Osterhaus, A.D.M.E.; Timens, W.; Turner, A.J.; Navis, G.; van Goor, H. The emerging role of ACE2 in physiology and disease. J. Pathol. 2007, 212, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.K.; Juno, J.A.; Lee, W.; Wragg, K.M.; Hogarth, P.M.; Kent, S.J.; Burrell, L.M. Plasma ACE2 activity is persistently elevated following SARS-CoV-2 infection: Implications for COVID-19 pathogenesis and consequences. Eur. Respir. J. 2021, 57, 2003730. [Google Scholar] [CrossRef]
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A.; Munier, C.M.L.; Patel, S.K.; Juno, J.A.; Burrell, L.M.; Kent, S.J.; Dore, G.J.; et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 210–216. [Google Scholar] [CrossRef]
- Úri, K.; Fagyas, M.; Kertész, A.; Borbély, A.; Jenei, C.; Bene, O.; Csanádi, Z.; Paulus, W.J.; Édes, I.; Papp, Z.; et al. Circulating ACE2 activity correlates with cardiovascular disease development. J. Renin. Angiotensin Aldosterone Syst. 2016, 17, 1470320316668435. [Google Scholar] [CrossRef]
- Úri, K.; Fagyas, M.; Mányiné Siket, I.; Kertész, A.; Csanádi, Z.; Sándorfi, G.; Clemens, M.; Fedor, R.; Papp, Z.; Édes, I.; et al. New perspectives in the Renin-Angiotensin-Aldosterone System (RAAS) IV: Circulating ACE2 as a biomarker of systolic dysfunction in human hypertension and heart failure. PLoS ONE 2014, 9, e87845. [Google Scholar] [CrossRef]
- Soro-Paavonen, A.; Gordin, D.; Forsblom, C.; Rosengard-Barlund, M.; Waden, J.; Thorn, L.; Sandholm, N.; Thomas, M.C.; Groop, P.H.; FinnDiane Study Group. Circulating ACE2 activity is increased in patients with type 1 diabetes and vascular complications. J. Hypertens. 2012, 30, 375–383. [Google Scholar] [CrossRef]
- Walters, T.E.; Kalman, J.; Patel, S.K.; Mearns, M.; Velkoska, E.; Burrell, L.M. Angiotensin converting enzyme 2 activity and human atrial fibrillation: Increased plasma angiotensin converting enzyme 2 activity is associated with atrial fibrillation and more advanced left atrial structural remodelling. Europace 2017, 19, 1280–1287. [Google Scholar] [CrossRef]
- Anguiano, L.; Riera, M.; Pascual, J.; Valdivielso, J.M.; Barrios, C.; Betriu, A.; Clotet, S.; Mojal, S.; Fernández, E.; Soler, M.J.; et al. Circulating angiotensin converting enzyme 2 activity as a biomarker of silent atherosclerosis in patients with chronic kidney disease. Athrerosclerosis 2016, 253, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.A.; Velkoska, E.; Ierino, F.L.; Burrell, L.M. Angiotensin-converting enzyme 2 activity in patients with chronic kidney disease. Nephrol. Dial. Transpl. 2013, 28, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Anguiano, L.; Riera, M.; Pascual, J.; Valdivielso, J.M.; Barrios, C.; Betriu, A.; Mojal, S.; Fernández, E.; Soler, M.J.; NEFRONA Study. Circulating angiotensin-converting enzyme 2 activity in patients with chronic kidney disease without previous history of cardiovascular disease. Nephrol. Dial. Transpl. 2015, 30, 1176–1185. [Google Scholar] [CrossRef]
- Narula, S.; Yusuf, S.; Chong, M.; Ramasundarahettige, C.; Rangarajan, S.; Bangdiwala, S.; van Eikels, M.; Leineweber, K.; Pigeyre, M.; Paré, G. Plasma ACE2 and risk of death or cardiometabolic diseases: A case-cohort analysis. Lancet 2020, 296, 968–976. [Google Scholar] [CrossRef]
- Ramchand, J.; Patel, S.; Srivastava, P.M.; Farouque, O.; Burrell, L.M. Elevated plasma angiotensin converting enzyme 2 activity is an independent predictor of major adverse cardiac events in patients with obstructive coronary artery disease. PLoS ONE 2018, 13, e0198144. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 180, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.; Arbous, M.; Gommers, D.; Kant, K.; Kaptein, F.; van Paassen, J.; Stals, M.; Huisman, M.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Juneja, G.K.; Castelo, M.; Yeh, C.H.; Cerroni, S.E.; Hansen, B.E.; Chessum, J.E.; Abraham, J.; Cani, E.; Dwivedi, D.J.; Fraser, D.D.; et al. Biomarkers of coagulation, endothelial function, and fibrinolysis in critically ill patients with COVID-19: A single-center prospective longitudinal study. J. Thromb. Haemost. 2021, 19, 1546–1557. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H.; Levi, M.; Thachil, J. Coagulopathy in COVID-19. J. Thromb. Haemost. 2021, 18, 2103–2109. [Google Scholar] [CrossRef]
- Lim, H.Y.; Donnan, G.; Nandurkar, H.; Ho, P. Global coagulation assays in hypercoagulable states. J. Thromb. Thrombolysis 2022. [CrossRef]
- Lim, H.Y.; O’Malley, C.; Donnan, G.; Nandurkar, H.; Ho, P. A review of global coagulation assays—Is there a role in thrombosis risk prediction? Thromb. Res. 2019, 179, 45–55. [Google Scholar] [CrossRef]
- Brummel, K.E.; Paradis, S.G.; Butenas, S.; Mann, K.G. Thrombin functions during tissue factor induced blood coagulation. Blood 2002, 100, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Lancé, M.D. A general review of major global coagulation assays: Thrombelastography, thrombin generation test and clot waveform analysis. Thromb. J. 2015, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hemker, H.C.; Al Dieri, R.; De Smedt, E.; Beguin, S. Thrombin generation, a function test of the haemostatic-thrombotic system. Thromb. Haemost. 2006, 96, 553–561. [Google Scholar]
- Rice, G.I.; Jones, A.L.; Grant, P.J.; Carter, A.M.; Turner, A.J.; Hooper, N.M. Circulating activities of angiotensin-converting enzyme, its homolog, angiotensin-converting enzyme 2, and neprilysin in a family study. Hypertension 2006, 48, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Lui, B.; Tacey, M.; Selan, C.; Donnan, G.; Burrell, L.M.; Nandurkar, H.; Ho, P. Global coagulation assays in healthy controls: Are there compensatory mechanisms within the coagulation system? J. Thromb. Thrombolysis 2021, 52, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Ramchand, J.; Patel, S.; Kearney, L.G.; Matalanis, G.; Farouque, O.; Srivastava, P.M.; Burrell, L.M. Plasma ACE2 activity predicts mortality in aortic stenosis and is associated with severe myocardial fibrosis. JACC Cardiovasc. Imaging 2020, 13, 655–664. [Google Scholar] [CrossRef]
- Hai, Q.; Smith, J.D. Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in cholesterol metabolism: From Its discovery to clinical trials and the genomics era. Metabolites 2021, 11, 543. [Google Scholar] [CrossRef]
- Hai, Q.; Ritchey, B.; Robinet, P.; Alzayed, A.M.; Brubaker, G.; Zhang, J.; Smith, J.D. Quantitative trait locus mapping of macrophage cholesterol metabolism and CRISPR/Cas9 editing Implicate an ACAT1 truncation as a causal modifier variant. Arter. Thromb. Vasc. Biol. 2018, 38, 83–91. [Google Scholar] [CrossRef]
- Melton, E.M.; Li, H.; Benson, J.; Sohn, P.; Huang, L.H.; Song, B.L.; Li, B.L.; Chang, C.C.Y.; Chang, T.Y. Myeloid Acat1/Soat1 KO attenuates pro-inflammatory responses in macrophages and protects against atherosclerosis in a model of advanced lesions. J. Biol. Chem. 2019, 294, 15836–15849. [Google Scholar] [CrossRef]
- Spiel, A.O.; Gilbert, J.C.; Jilma, B. Von Willebrand Factor in cardiovascular disease. Circulation 2008, 117, 1449–1459. [Google Scholar] [CrossRef]
- Ritchey, B.; Hai, Q.; Han, J.; Barnard, J.; Smith, J.D. Genetic variant in 3′ untranslated region of the mouse pycard gene regulates inflammasome activity. eLife 2021, 10, e68203. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Shen, D.L.; Zhu, K.; Tang, J.N.; Hai, Q.M.; Zhang, J.Y. Levels of interleukin-33 and interleukin-6 in patients with acute coronary syndrome or stable angina. Clin. Investig. Med. 2013, 36, E234–E241. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Burrell, L.M.; Brook, R.; Nandurkar, H.H.; Donnan, G.; Ho, P. The need for individualized risk assessment in cardiovascular disease. J. Pers. Med. 2022, 12, 1140. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.G.; Isermann, B.; Kleber, M.E.; Wang, H.; Boehm, B.O.; Grammer, T.B.; Prueller, F.; Nawroth, P.P.; Maerz, W. Inverse association of the endogenous thrombin potential (ETP) with cardiovascular death: The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Int. J. Cardiol. 2014, 176, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Winckers, K.; ten Cate, H.; Hackeng, T.M. The role of tissue factor pathway inhibitor in atherosclerosis and arterial thrombosis. Blood Rev. 2013, 27, 119–132. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, R.B., Sr.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General cardiovascular risk profile for use in primary care: The Framingham Heart Study. Circulation 2008, 117, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef]
- Thatcher, S.E.; Zhang, X.; Howatt, D.A.; Lu, H.; Gurley, S.B.; Daugherty, A.; Cassis, L.A. Angiotensin-converting enzyme 2 deficiency in whole body or bone marrow-derived cells increases atherosclerosis in low-density lipoprotein receptor -/- mice. Arter. Thromb. Vasc. Biol. 2011, 31, 758–765. [Google Scholar] [CrossRef]
- Wu, C.; Mohammadmoradi, S.; Chen, J.; Sawada, H.; Daugherty, A.; Lu, H. Renin-angiotensin system and cardiovascular functions. Arter. Thromb. Vasc. Biol. 2018, 38, e108–e116. [Google Scholar] [CrossRef]
- Dong, B.; Zhang, C.; Feng, J.B.; Zhao, Y.X.; Li, S.Y.; Yang, Y.P.; Dong, Q.L.; Deng, B.P.; Zhu, L.; Yu, Q.T.; et al. Overexpression of ACE2 enhances plaque stability in a rabbit model of atherosclerosis. Arter. Thromb. Vasc. Biol. 2008, 28, 1270–1276. [Google Scholar] [CrossRef]
- Wang, Y.; Tikellis, C.; Thomas, M.C.; Golledge, J. Angiotensin converting enzyme 2 and atherosclerosis. Atherosclerosis 2013, 226, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.M.; Franca-Falcao, M.S.; Calzerra, N.T.M.; Luz, M.S.; Gadelha, D.D.A.; Balarini, C.M.; Queiroz, T.M. Role of renin-angiotensin system components in atherosclerosis: Focus on Ang-II, ACE2, and Ang-1-7. Front Physiol. 2020, 11, 1067. [Google Scholar] [CrossRef]
- Granger, D.N.; Rodrigues, S.F.; Yildirim, A.; Senchenkova, E.Y. Microvascular responses to cardiovascular risk factors. Microcirculation 2010, 17, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.; Berne, M.; Somasundaran, M.; Sullivan, J.; Luzuriaga, K.; Grrenough, T.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]

{kind=link}
| All Controls | Undetectable Plasma ACE2 Activity | Detectable Plasma ACE2 Activity | p-Value | |
|---|---|---|---|---|
| N | 123 | 59 (48.0%) | 64 (52.0%) | |
| Age, mean (SD) | 41 (17) | 40 (17) | 43 (17) | 0.40 |
| Sex (male) | 43 (35.0%) | 43 (72.9%) | 43 (67.2%) | 0.29 |
| Plasma ACE2 activity (pmol/mL/min) | 0.10 (0.02, 3.33) | 0.02 | 3.20 (1.17, 7.82) | |
| Baseline investigations | ||||
| Haemoglobin (g/dL) | 142.0 (136.0, 153.0) | 141.0 (135.0, 153.0) | 142.0 (137.0, 153.0) | 0.38 |
| White cell count (×109/L) | 5.7 (5.0, 6.6) | 6.1 (5.3, 6.8) | 5.6 (4.8, 6.3) | 0.031 |
| Platelets (×109/L) | 245.0 (215.0, 279.0) | 257.0 (229.0, 293.0) | 230.0 (202.0, 272.0) | 0.006 |
| Prothrombin time (sec) | 11.0 (10.3, 12.6) | 12.2 (11.0, 13.2) | 10.9 (10.1, 11.0) | <0.001 |
| APTT (sec) | 28.0 (26.0, 31.0) | 28.8 (26.4, 31.6) | 27.9 (25.0, 30.2) | 0.062 |
| Fibrinogen (g/L) | 3.0 (2.5, 3.5) | 3.1 (2.6, 3.5) | 2.9 (2.5, 3.5) | 0.12 |
| D-dimer (mg/L FEU) | 270.0 (150.0, 290.0) | 270.0 (190.0, 300.0) | 190.0 (150.0, 270.0) | 0.035 |
| Factor VIII level (%) | 104.0 (86.0, 145.0) | 111.0 (86.0, 151.0) | 102.0 (84.0, 130.5) | 0.098 |
| Von Willebrand factor antigen (%) | 100.0 (86.0, 127.0) | 141.0 (135.0, 153.0) | 142.0 (137.0, 153.0) | 0.38 |
| eGFR (mL/min/1.73 m2) | 99.5 (90.0, 116.0) | 103.0 (89.0, 117.0) | 99.0 (91.0, 113.0) | 0.67 |
| Glycated haemoglobin, HbA1c (%) | 5.4 (5.1, 5.5) | 5.2 (5.1, 5.5) | 5.4 (5.1, 5.7) | 0.072 |
| Total cholesterol (mmol/L) | 5.1 (4.4, 6.0) | 5.0 (4.4, 5.5) | 5.3 (4.2, 6.1) | 0.35 |
| High density lipoprotein (HDL) (mmol/L) | 1.5 (1.3, 1.9) | 1.5 (1.3, 1.8) | 1.6 (1.4, 2.0) | 0.085 |
| Low density lipoprotein (LDL) (mmol/L) | 3.0 (2.3, 3.6) | 2.9 (2.6, 3.5) | 3.1 (2.2, 3.6) | 0.94 |
| Triglycerides (mmol/L) | 1.0 (0.7, 1.4) | 1.0 (0.7, 1.5) | 1.0 (0.7, 1.4) | 0.93 |
| Thromboelastography (TEG) | ||||
| R-time (min) | 6.3 (5.4, 7.9) | 6.3 (5.4, 7.2) | 6.5 (5.6, 8.2) | 0.30 |
| K-time (min) | 2.2 (1.8, 2.6) | 2.1 (1.6, 2.4) | 2.3 (1.8, 2.8) | 0.49 |
| a-angle (°) | 58.1 (48.1, 64.9) | 58.5 (49.4, 65.0) | 57.6 (46.9, 63.4) | 0.70 |
| Maximum Amplitude (mm), mean (SD) | 59.9 (6.1) | 62.0 (5.5) | 57.4 (5.9) | <0.001 |
| Lysis 30 (%) | 0.6 (0.0, 1.5) | 0.6 (0.1, 1.3) | 0.6 (0.0, 2.6) | 0.61 |
| Calibrated automated Thrombogram (CAT) | ||||
| Lag time (min) | 3.1 (2.7, 3.7) | 3.1 (2.7, 3.6) | 3.3 (2.7, 3.7) | 0.41 |
| Endogenous thrombin potential (nM.min) | 1349.0 (1176.0, 1552.0) | 1360.1 (1180.8, 1528.2) | 1336.0 (1151.5, 1590.0) | 0.83 |
| Peak thrombin (nM) | 227.0 (177.0, 272.3) | 233.5 (177.2, 273.7) | 221.7 (174.3, 271.1) | 0.45 |
| Velocity Index (nM/min) | 68.7 (46.2, 99.9) | 74.1 (46.2, 100.2) | 68.4 (46.6, 99.1) | 0.81 |
| Overall Haemostasis Potential (OHP) | ||||
| Overall coagulation profile, mean (SD) | 36.1 (9.8) | 34.3 (9.7) | 37.8 (9.6) | 0.052 |
| Overall haemostasis potential (OHP) | 6.5 (4.8, 9.7) | 6.2 (4.7, 9.5) | 6.7 (5.2, 9.9) | 0.30 |
| Overall fibrinolytic potential (OFP) (%) | 80.9 (77.2, 84.2) | 80.5 (76.9, 84.2) | 81.3 (78.3, 84.2) | 0.51 |
| Tissue factor pathway inhibitor (ng/mL) | 12.7 (66.5, 26.7) | 10.4 (6.5, 24.1) | 16.6 (6.7, 27.6) | 0.30 |
| Factor | Healthy Controls | Patients with Cardiovascular Risk Factors and/or Disease | p-Value |
|---|---|---|---|
| N | 123 | 258 | |
| Age, mean (SD) | 41.5 (16.6) | 64.5 (13.9) | <0.001 |
| Sex (male) | 43 (35.0%) | 142 (55%) | <0.001 |
| Plasma ACE2 activity (pmol/mL/min) | 0.10 (0.02, 3.33) | 5.99 (1.95, 10.37) | <0.001 |
| Comorbidities | |||
| Obesity, n (%) | Data not collected | 136 (52.7%) | |
| Hypertension, n (%) | 0 | 212 (82.2%) | |
| Diabetes mellitus, n (%) | 0 | 156 (60.5%) | |
| Cerebrovascular accident, n (%) | 0 | 16 (6.2%) | |
| Peripheral artery disease, n (%) | 0 | 18 (7.0%) | |
| Coronary artery disease, n (%) | 0 | 72 (27.9%) | |
| Active smoker, n (%) | 0 | 45 (17.4%) | |
| Number of cardiovascular risk factors, mean (SD) | 0 | 4.5 (1.5) | |
| Medications | |||
| Aspirin, n (%) | 0 | 106 (41.1%) | |
| Statin, n (%) | 0 | 170 (65.9%) | |
| ACE inhibitor or ARB, n (%) | 0 | 143 (55.4%) | |
| Biochemical investigations | |||
| Haemoglobin (g/dL) | 142.0 (136.0, 153.0) | 135.5 (119.0, 147.0) | <0.001 |
| White cell count (×109/L) | 5.7 (5.0, 6.6) | 7.3 (6.2, 9.0) | <0.001 |
| Platelets (×109/L) | 245.0 (215.0, 279.0) | 249.5 (195.8, 290.1) | 0.82 |
| Prothrombin time (s) | 11.0 (10.3, 12.6) | 12.0 (10.9, 12.8) | <0.001 |
| Activated partial thromboplastin time (s) | 28.0 (26.0, 31.0) | 28.1 (26.5, 29.9) | 0.84 |
| Fibrinogen (g/L) | 3.0 (2.5, 3.5) | 3.9 (3.3, 4.6) | <0.001 |
| D-dimer (mg/L FEU) | 270.0 (150.0, 290.0) | 570.0 (340.0, 980.0) | <0.001 |
| Factor VIII level (%) | 104.0 (86.0, 145.0) | 161.0 (118.5, 198.0) | <0.001 |
| Von Willebrand factor antigen (%) | 100.0 (86.0, 127.0) | 154.0 (112.0, 193.3) | <0.001 |
| Estimated glomerular filtration rate (mL/min/1.73 m2) | 99.5 (90.0, 116.0) | 67.0 (30.0, 90.0) | <0.001 |
| Glycated haemoglobin, HbA1c (%) | 5.4 (5.1, 5.5) | 6.4 (5.5, 7.6) | <0.001 |
| Total Cholesterol (mmol/L) | 5.1 (4.4, 6.0) | 4.3 (3.6, 5.1) | <0.001 |
| High density lipoprotein (HDL) (mmol/L) | 1.5 (1.3, 1.9) | 1.1 (0.9, 1.4) | <0.001 |
| Low density lipoprotein (LDL) (mmol/L) | 3.0 (2.3, 3.6) | 2.2 (1.6, 2.9) | <0.001 |
| Triglycerides (mmol/L) | 1.0 (0.7, 1.4) | 1.8 (1.2, 2.5) | <0.001 |
| Thromboelastography (TEG) | |||
| R-time (min) | 6.3 (5.4, 7.9) | 6.2 (5.3, 7.2) | 0.050 |
| K-time (min) | 2.2 (1.8, 2.6) | 1.8 (1.4, 2.1) | <0.001 |
| a-angle (°) | 58.1 (48.1, 64.9) | 59.2 (47.8, 68.0) | 0.29 |
| Maximum Amplitude, mean (SD) | 59.9 (6.1) | 69.2 (5.9) | <0.001 |
| Lysis 30 (%) | 0.6 (0.0, 1.5) | 0.1 (0.0, 0.6) | <0.001 |
| Calibrated automated thrombogram (CAT) | |||
| Lag time (min) | 3.1 (2.7, 3.7) | 4.0 (3.4, 4.7) | <0.001 |
| Endogenous thrombin potential (nM.min) | 1349.0 (1176.0, 1552.0) | 1310.9 (1132.4, 1482.6) | 0.17 |
| Peak thrombin (nM) | 227.0 (177.0, 272.3) | 221.5 (178.7, 264.9) | 0.87 |
| Velocity Index (nM/min) | 68.7 (46.2, 99.9) | 64.3 (42.3, 92.7) | 0.63 |
| Overall haemostasis potential (OHP) | |||
| Overall coagulation profile, mean (SD) (unit) | 36.1 (9.8) | 42.2 (10.5) | <0.001 |
| Overall haemostasis potential (OHP) | 6.5 (4.8, 9.7) | 9.8 (7.0, 14.4) | <0.001 |
| Overall fibrinolytic potential (OFP) (%) | 80.9 (77.2, 84.2) | 74.8 (68.9, 80.2) | <0.001 |
| Tissue factor pathway inhibitor (ng/mL) | 12.7 (66.5, 26.7) | 35.2 (17.8, 55.4) | <0.001 |
| Plasma ACE2 Activity ≤3 pmol/mL/min | Plasma ACE2 Activity 3.1 to 8.2 pmol/mL/min | Plasma ACE2 Activity ≥8.3 pmol/mL/min | p-Value 1 | p-Value 2 | |
|---|---|---|---|---|---|
| N | 85 | 86 | 87 | ||
| Age, mean (SD) | 60.5 (15.9) | 64.9 (12.7) | 68.2 (12.0) | <0.001 | |
| Sex (male) | 35 (41%) | 46 (54%) | 61 (70%) | 0.002 | |
| Clinical characteristics | |||||
| Body mass index (kg/m2), mean (SD) | 30.8 (8.3) | 31.7 (7.9) | 31.8 (7.8) | 0.65 | |
| Systolic blood pressure (mmHg), mean (SD) | 133 (16) | 136 (18) | 136 (20) | 0.54 | |
| Diastolic blood pressure (mmHg), mean (SD) | 77 (11) | 76 (12) | 75 (13) | 0.69 | |
| Framingham Grade | <0.001 | <0.001 | |||
| Low (<10) | 24 (28.2%) | 9 (10.4%) | 8 (9.2%) | ||
| Moderate (10–20) | 23 (27.1%) | 29 (33.7%) | 12 (13.8%) | ||
| High (>20) | 38 (44.7%) | 48 (55.8%) | 67 (77.0%) | ||
| Comorbidities | |||||
| Obesity | 38 (44.7%) | 50 (58.1%) | 48 (55.2%) | 0.18 | |
| Hypertension | 70 (82.4%) | 73 (84.9%) | 69 (79.3%) | 0.63 | |
| Diabetes mellitus | 48 (56.5%) | 48 (55.8%) | 60 (69.0%) | 0.14 | |
| Cerebrovascular accident | 4 (4.8%) | 7 (8.1%) | 5 (5.7%) | 0.64 | |
| Peripheral artery disease | 3 (3.5%) | 5 (5.8%) | 10 (11.5%) | 0.11 | |
| Coronary artery disease | 15 (17.6%) | 24 (27.9%) | 33 (37.9%) | 0.012 | 0.005 |
| Active smoker | 19 (22.4%) | 13 (15.1%) | 13 (14.9%) | 0.35 | |
| Number of cardiovascular risk factors 3, mean (SD) | 4.1 (1.7) | 4.4 (1.3) | 4.8 (1.2) | 0.003 | |
| Medications | |||||
| Aspirin | 26 (30.6%) | 38 (44.2%) | 42 (48.3%) | 0.048 | |
| Statin | 53 (62.4%) | 58 (68.2%) | 59 (67.8%) | 0.66 | |
| ACE inhibitor or ARB | 42 (49.4%) | 49 (57.0%) | 52 (59.8%) | 0.72 | |
| Biochemical Investigations | |||||
| Haemoglobin (g/dL) | 133.5 (118.8, 145.3) | 135.5 (118.8, 148.3) | 136.0 (120.0, 147.3) | 0.61 | |
| White cell count (×109/L) | 7.8 (6.1, 9.1) | 7.4 (6.3, 8.8) | 7.2 (5.9, 9.0) | 0.75 | |
| Platelets (×109/L) | 264.5 (198.5, 313.8) | 246.5 (197.0, 288.5) | 229.0 (171.8, 288.3) | 0.063 | |
| Prothrombin time (s) | 11.4 (10.7, 12.8) | 12.1 (11.0, 12.5) | 12.3 (11.4, 13.2) | 0.001 | 0.044 |
| Activated partial thromboplastin time (s) | 28.4 (26.2, 29.5) | 27.9 (26.7, 30.1) | 28.1 (26.4, 30.4) | 0.53 | |
| Fibrinogen (g/L) | 3.9 (3.2, 4.5) | 4.2 (3.4, 4.9) | 3.7 (3.2, 4.5) | 0.16 | |
| D-dimer (mg/L FEU) | 540.0 (300.0, 955.0) | 520.0 (320.0, 840.0) | 610.0 (340.0, 1030.0) | 0.42 | |
| Factor VIII level (%) | 153.5 (114.0, 195.0) | 158.5 (114.0, 188.0) | 174.5 (130.0, 207.3) | 0.072 | |
| Von Willebrand factor antigen (%) | 150.0 (110.0, 183.0) | 147.0 (102.5, 194.0) | 169.0 (122.0, 216.0) | 0.096 | |
| C-reactive protein (mg/L) | 3.0 (1.0, 8.5) | 2.9 (1.3, 6.8) | 3.2 (1.2, 6.4) | 0.99 | |
| Estimated glomerular filtration rate (mL/min/1.73 m2) | 65.0 (19.0, 93.5) | 72.0 (38.0, 91.0) | 61.0 (30.5, 87.0) | 0.66 | |
| Glycated haemoglobin, HbA1c(%) | 6.1 (5.4, 7.6) | 6.3 (5.5, 7.6) | 6.4 (5.7, 7.8) | 0.18 | |
| Total cholesterol (mmol/L) | 4.4 (3.8, 5.4) | 4.3 (3.5, 5.0) | 4.1 (3.4, 5.0) | 0.13 | |
| High density lipoprotein (HDL) (mmol/L) | 1.2 (0.9, 1.5) | 1.1 (0.9, 1.5) | 1.1 (1.0, 1.4) | 0.87 | |
| Low density lipoprotein (LDL) (mmol/L) | 2.5 (1.8, 3.2) | 2.2 (1.5, 2.8) | 2.0 (1.6, 2.7) | 0.051 | |
| Triglycerides (mmol/L) | 1.6 (1.2, 2.4) | 1.7 (1.2, 2.4) | 1.9 (1.2, 2.7) | 0.72 | |
| Plasma ACE2 Activity ≤3 pmol/mL/min | Plasma ACE2 Activity 3.1 to 8.2 pmol/mL/min | Plasma ACE2 Activity ≥8.3 pmol/mL/min | p-Value 1 | p-Value 2 | |
|---|---|---|---|---|---|
| N | 85 | 86 | 87 | ||
| Thromboelastography | |||||
| R-time (min) | 6.2 (5.3, 7.6) | 6.1 (5.2, 7.5) | 6.2 (5.4, 7.0) | 0.99 | |
| K-time (min) | 1.8 (1.3, 2.2) | 1.8 (1.4, 2.2) | 1.7 (1.4, 2.1) | 0.95 | |
| a-angle (°) | 56.4 (46.3, 67.9) | 54.0 (46.6, 68.2) | 62.4 (50.3, 69.1) | 0.28 | |
| Maximum Amplitude (mm), mean (SD) | 69.2 (5.6) | 68.7 (5.6) | 66.1 (6.2) | 0.009 | 0.043 |
| Lysis 30 (%) | 0.1 (0.0, 0.9) | 0.1 (0.0, 0.7) | 0.1 (0.0, 0.5) | 0.70 | |
| Calibrated Automated Thrombogram | |||||
| Lag time (min) | 4.0 (3.4, 4.7) | 3.7 (3.3, 4.3) | 4.3 (3.7, 5.2) | 0.025 | 0.094 |
| Endogenous thrombin potential (nM.min) | 1272.2 (1148.1, 1449.6) | 1307.5 (1123.2, 1525.2) | 1305.1 (1080.5, 1471.1) | 0.87 | |
| Peak thrombin (nM) | 217.0 (176.3, 254.6) | 225.2 (177.1, 296.4) | 227.8 (182.3, 260.6) | 0.55 | |
| Velocity Index (nM/min) | 63.1 (41.4, 85.5) | 67.6 (47.7, 110.1) | 64.4 (42.4, 93.6) | 0.25 | |
| Overall Haemostasis Potential | |||||
| Overall coagulation profile, mean (SD) (unit) | 42.8 (11.8) | 42.1 (9.9) | 38.9 (9.5) | 0.035 | 0.086 |
| Overall haemostasis potential (OHP) | 10.7 (6.5, 15.6) | 10.5 (7.6, 14.6) | 9.2 (6.6, 12.7) | 0.51 | |
| Overall fibrinolytic potential (OFP) (%) | 75.2 (67.7, 80.4) | 74.7 (67.6, 79.6) | 74.9 (68.8, 81.2) | 0.95 | |
| Tissue factor pathway inhibitor (ng/mL) | 37.4 (19.1, 56.7) | 26.9 (13.5, 46.3) | 36.3 (17.1, 77.1) | 0.027 | 0.044 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, H.Y.; Patel, S.K.; Huang, P.; Tacey, M.; Choy, K.W.; Wang, J.; Donnan, G.; Nandurkar, H.H.; Ho, P.; Burrell, L.M. Plasma Angiotensin Converting Enzyme 2 (ACE2) Activity in Healthy Controls and Patients with Cardiovascular Risk Factors and/or Disease. J. Pers. Med. 2022, 12, 1495. https://doi.org/10.3390/jpm12091495
Lim HY, Patel SK, Huang P, Tacey M, Choy KW, Wang J, Donnan G, Nandurkar HH, Ho P, Burrell LM. Plasma Angiotensin Converting Enzyme 2 (ACE2) Activity in Healthy Controls and Patients with Cardiovascular Risk Factors and/or Disease. Journal of Personalized Medicine. 2022; 12(9):1495. https://doi.org/10.3390/jpm12091495
Chicago/Turabian StyleLim, Hui Yin, Sheila K. Patel, Ping Huang, Mark Tacey, Kay Weng Choy, Julie Wang, Geoffrey Donnan, Harshal H. Nandurkar, Prahlad Ho, and Louise M. Burrell. 2022. "Plasma Angiotensin Converting Enzyme 2 (ACE2) Activity in Healthy Controls and Patients with Cardiovascular Risk Factors and/or Disease" Journal of Personalized Medicine 12, no. 9: 1495. https://doi.org/10.3390/jpm12091495