
Cell-Free Fetal DNA and Non-Invasive Prenatal Diagnosis of Chromosomopathies and Pediatric Monogenic Diseases: A Critical Appraisal and Medicolegal Remarks

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Eligibility Criteria
- Language: studies written in English.
- Study design: no restriction.
- We excluded from this review any source exhibiting insufficient data to elaborate and report results.
2.3. Information Sources and Search Strategy
2.4. Study Selection and Data Extraction
3. Results
3.1. Non-Invasive Prenatal Diagnosis of Chromosomal Aneuploidies
3.2. Non-Invasive Prenatal Diagnosis of CNV Diseases
3.3. Non-Invasive Prenatal Diagnosis of Monogenic Transmission Diseases
3.3.1. Endocrine System and Bone Diseases
3.3.2. Metabolic Disorders
3.3.3. Neuromuscular Pathologies
3.3.4. Hematologic Disorders
3.3.5. Skin Diseases
3.3.6. Other Conditions (Wilson Disease, Cystic Fibrosis, Non-Syndromic Hearing Loss, Polycystic Kidney Disease, 46XY Sex Development Disorders)
3.4. Causes of False Positives and False Negatives in cffDNA Test
4. Discussion
Legal and Ethical Implications Call for Caution
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scotchman, E.; Shaw, J.; Paternoster, B.; Chandler, N.; Chitty, L.S. Non-Invasive Prenatal Diagnosis and Screening for Monogenic Disorders. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 253, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, M.; Zhu, Y.; Jiang, L.; Li, J.; Wang, Y.; Lu, Z.; Guo, F.; Wang, H.; Peng, Z.; et al. Noninvasive Prenatal Diagnosis of Monogenic Disorders Based on Direct Haplotype Phasing through Targeted Linked-Read Sequencing. BMC Med. Genom. 2021, 14, 244. [Google Scholar] [CrossRef] [PubMed]
- Tamminga, S.; van Maarle, M.; Henneman, L.; Oudejans, C.B.M.; Cornel, M.C.; Sistermans, E.A. Maternal Plasma DNA and RNA Sequencing for Prenatal Testing. Adv. Clin. Chem. 2016, 74, 63–102. [Google Scholar] [CrossRef] [PubMed]
- Judah, H.; Gil, M.M.; Syngelaki, A.; Galeva, S.; Jani, J.; Akolekar, R.; Nicolaides, K.H. Cell-Free DNA Testing of Maternal Blood in Screening for Trisomies in Twin Pregnancy: Updated Cohort Study at 10-14 Weeks and Meta-Analysis. Ultrasound Obstet. Gynecol. 2021, 58, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Borth, H.; Teubert, A.; Glaubitz, R.; Knippenberg, S.; Kutur, N.; Winkler, T.; Eiben, B. Analysis of Cell-Free DNA in a Consecutive Series of 13,607 Routine Cases for the Detection of Fetal Chromosomal Aneuploidies in a Single Center in Germany. Arch. Gynecol. Obstet. 2021, 303, 1407–1414. [Google Scholar] [CrossRef]
- Serapinas, D.; Boreikaitė, E.; Bartkevičiūtė, A.; Norvilaitė, K.; Narbekovas, A.; Bartkevičienė, D. The Level of Free Fetal DNA as Precise Noninvasive Marker for Chromosomal Aneuploidies: First Results from BALTIC Region. Medicina 2020, 56, 579. [Google Scholar] [CrossRef]
- Gil, M.M.; Galeva, S.; Jani, J.; Konstantinidou, L.; Akolekar, R.; Plana, M.N.; Nicolaides, K.H. Screening for Trisomies by CfDNA Testing of Maternal Blood in Twin Pregnancy: Update of The Fetal Medicine Foundation Results and Meta-Analysis. Ultrasound Obstet. Gynecol. 2019, 53, 734–742. [Google Scholar] [CrossRef] [Green Version]
- Miltoft, C.B.; Rode, L.; Ekelund, C.K.; Sundberg, K.; Kjaergaard, S.; Zingenberg, H.; Tabor, A. Contingent First-Trimester Screening for Aneuploidies with Cell-Free DNA in a Danish Clinical Setting. Ultrasound Obstet. Gynecol. 2018, 51, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Gil, M.M.; Accurti, V.; Santacruz, B.; Plana, M.N.; Nicolaides, K.H. Analysis of Cell-Free DNA in Maternal Blood in Screening for Aneuploidies: Updated Meta-Analysis. Ultrasound Obstet. Gynecol. 2017, 50, 302–314. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Phillips, S.; Freeman, K.; Geppert, J.; Agbebiyi, A.; Uthman, O.A.; Madan, J.; Clarke, A.; Quenby, S.; Clarke, A. Accuracy of Non-Invasive Prenatal Testing Using Cell-Free DNA for Detection of Down, Edwards and Patau Syndromes: A Systematic Review and Meta-Analysis. BMJ Open 2016, 6, e010002. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.; Jiang, F.; Fu, M.; Yuan, Y.; Guo, Y.; Zhu, Z.; Lin, M.; Liu, Q.; Tian, Z.; et al. Non-Invasive Prenatal Testing for Trisomies 21, 18 and 13: Clinical Experience from 146 958 Pregnancies: Clinical Experience of NIPT from 146 958 Pregnancies. Ultrasound Obstet. Gynecol. 2015, 45, 530–538. [Google Scholar] [CrossRef]
- Porreco, R.P.; Garite, T.J.; Maurel, K.; Marusiak, B.; Obstetrix Collaborative Research Network; Ehrich, M.; van den Boom, D.; Deciu, C.; Bombard, A. Noninvasive Prenatal Screening for Fetal Trisomies 21, 18, 13 and the Common Sex Chromosome Aneuploidies from Maternal Blood Using Massively Parallel Genomic Sequencing of DNA. Am. J. Obstet. Gynecol. 2014, 211, 365.e1–365.e12. [Google Scholar] [CrossRef]
- Lau, T.K.; Cheung, S.W.; Lo, P.S.S.; Pursley, A.N.; Chan, M.K.; Jiang, F.; Zhang, H.; Wang, W.; Jong, L.F.J.; Yuen, O.K.C.; et al. Non-Invasive Prenatal Testing for Fetal Chromosomal Abnormalities by Low-Coverage Whole-Genome Sequencing of Maternal Plasma DNA: Review of 1982 Consecutive Cases in a Single Center. Ultrasound Obstet. Gynecol. 2014, 43, 254–264. [Google Scholar] [CrossRef]
- Stumm, M.; Entezami, M.; Haug, K.; Blank, C.; Wüstemann, M.; Schulze, B.; Raabe-Meyer, G.; Hempel, M.; Schelling, M.; Ostermayer, E.; et al. Diagnostic Accuracy of Random Massively Parallel Sequencing for Non-Invasive Prenatal Detection of Common Autosomal Aneuploidies: A Collaborative Study in Europe. Prenat. Diagn. 2014, 34, 185–191. [Google Scholar] [CrossRef]
- Liang, D.; Lv, W.; Wang, H.; Xu, L.; Liu, J.; Li, H.; Hu, L.; Peng, Y.; Wu, L. Non-Invasive Prenatal Testing of Fetal Whole Chromosome Aneuploidy by Massively Parallel Sequencing. Prenat. Diagn. 2013, 33, 409–415. [Google Scholar] [CrossRef]
- Bardi, F.; Bosschieter, P.; Verheij, J.; Go, A.; Haak, M.; Bekker, M.; Sikkel, E.; Coumans, A.; Pajkrt, E.; Bilardo, C. Is There Still a Role for Nuchal Translucency Measurement in the Changing Paradigm of First Trimester Screening? Prenat. Diagn. 2020, 40, 197–205. [Google Scholar] [CrossRef]
- Lv, W.; Liang, L.; Chen, X.; Li, Z.; Liang, D.; Zhu, H.; Teng, Y.; Wu, W.; Wu, L.; Han, L. Noninvasive Prenatal Testing of Methylmalonic Acidemia CblC Type Using the CSMART Assay for MMACHC Gene Mutations. Front. Genet. 2021, 12, 750719. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, X.; Liu, L.; Dai, P.; Kong, X. Noninvasive Prenatal Diagnosis of Duchenne Muscular Dystrophy in Five Chinese Families Based on Relative Mutation Dosage Approach. BMC Med. Genom. 2021, 14, 275. [Google Scholar] [CrossRef]
- Lv, W.; Linpeng, S.; Li, Z.; Liang, D.; Jia, Z.; Meng, D.; Cram, D.S.; Zhu, H.; Teng, Y.; Yin, A.; et al. Noninvasive Prenatal Diagnosis for Pregnancies at Risk for β-Thalassaemia: A Retrospective Study. BJOG 2021, 128, 448–457. [Google Scholar] [CrossRef]
- Kong, L.; Li, S.; Zhao, Z.; Feng, J.; Chen, G.; Liu, L.; Tang, W.; Li, S.; Li, F.; Han, X.; et al. Haplotype-Based Noninvasive Prenatal Diagnosis of 21 Families With Duchenne Muscular Dystrophy: Real-World Clinical Data in China. Front. Genet. 2021, 12, 791856. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Tang, Y.-A.; Lee, I.-W.; Chang, F.-M.; Chien, C.-W.; Pan, H.-A.; Sun, H.S. Development and Validation of an Expanded Targeted Sequencing Panel for Non-Invasive Prenatal Diagnosis of Sporadic Skeletal Dysplasia. BMC Med. Genom. 2021, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Morshneva, A.; Kozyulina, P.; Vashukova, E.; Tarasenko, O.; Dvoynova, N.; Chentsova, A.; Talantova, O.; Koroteev, A.; Ivanov, D.; Serebryakova, E.; et al. Pilot Screening of Cell-Free MtDNA in NIPT: Quality Control, Variant Calling, and Haplogroup Determination. Genes 2021, 12, 743. [Google Scholar] [CrossRef] [PubMed]
- De Falco, L.; Piscopo, C.; D’Angelo, R.; Evangelista, E.; Suero, T.; Sirica, R.; Ruggiero, R.; Savarese, G.; Di Carlo, A.; Furino, G.; et al. Detection of 46, XY Disorder of Sex Development (DSD) Based on Plasma Cell-Free DNA and Targeted Next-Generation Sequencing. Genes 2021, 12, 1890. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ye, Y.; Fan, D.; Lin, S.; Li, M.; Hou, H.; Zhang, J.; Yang, X. Non-invasive Prenatal Diagnosis of Thalassemia through Multiplex PCR, Target Capture and Next-generation Sequencing. Mol. Med. Rep. 2020, 22, 1547–1557. [Google Scholar] [CrossRef]
- Jang, S.S.; Lim, B.C.; Yoo, S.-K.; Shin, J.-Y.; Kim, K.-J.; Seo, J.-S.; Kim, J.-I.; Chae, J.H. Targeted Linked-Read Sequencing for Direct Haplotype Phasing of Maternal DMD Alleles: A Practical and Reliable Method for Noninvasive Prenatal Diagnosis. Sci. Rep. 2018, 8, 8678. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Du, Y.; Zhang, H.; Wang, Z.; Wang, J.; Fu, X.; Cui, Y.; Chen, C.; Liang, J.; Xuan, Z.; et al. Identification of a de Novo Fetal Variant in Osteogenesis Imperfecta by Targeted Sequencing-Based Noninvasive Prenatal Testing. J. Hum. Genet. 2018, 63, 1129–1137. [Google Scholar] [CrossRef]
- Bijarnia-Mahay, S.; Häberle, J.; Jalan, A.B.; Puri, R.D.; Kohli, S.; Kudalkar, K.; Rüfenacht, V.; Gupta, D.; Maurya, D.; Verma, J.; et al. Urea Cycle Disorders in India: Clinical Course, Biochemical and Genetic Investigations, and Prenatal Testing. Orphanet. J. Rare Dis. 2018, 13, 174. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Chen, C.; Yuan, Y.; Han, L.; Wang, Y.; Qiu, W.; Zhang, H.; Asan; Gu, X. Haplotype-Based Noninvasive Prenatal Diagnosis of Hyperphenylalaninemia through Targeted Sequencing of Maternal Plasma. Sci. Rep. 2018, 8, 161. [Google Scholar] [CrossRef] [Green Version]
- Parks, M.; Court, S.; Bowns, B.; Cleary, S.; Clokie, S.; Hewitt, J.; Williams, D.; Cole, T.; MacDonald, F.; Griffiths, M.; et al. Non-Invasive Prenatal Diagnosis of Spinal Muscular Atrophy by Relative Haplotype Dosage. Eur. J. Hum. Genet. 2017, 25, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Li, Z.; Wang, W.; Huang, S.; Lu, Y.; Gao, Z.; Wang, L.; Kang, D.; Li, L.; Liu, Y.; et al. A Quantitative CSMART Assay for Noninvasive Prenatal Screening of Autosomal Recessive Nonsyndromic Hearing Loss Caused by GJB2 and SLC26A4 Mutations. Genet. Med. 2017, 19, 1309–1316. [Google Scholar] [CrossRef]
- Vermeulen, C.; Geeven, G.; de Wit, E.; Verstegen, M.J.A.M.; Jansen, R.P.M.; van Kranenburg, M.; de Bruijn, E.; Pulit, S.L.; Kruisselbrink, E.; Shahsavari, Z.; et al. Sensitive Monogenic Noninvasive Prenatal Diagnosis by Targeted Haplotyping. Am. J. Hum. Genet. 2017, 101, 326–339. [Google Scholar] [CrossRef]
- Ma, D.; Yuan, Y.; Luo, C.; Wang, Y.; Jiang, T.; Guo, F.; Zhang, J.; Chen, C.; Sun, Y.; Cheng, J.; et al. Noninvasive Prenatal Diagnosis of 21-Hydroxylase Deficiency Using Target Capture Sequencing of Maternal Plasma DNA. Sci. Rep. 2017, 7, 7427. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Y.; Wang, B.; Mao, J.; Wang, T.; Ye, K.; Ye, Y.; Cram, D.S.; Li, H. Development and Validation of a Fetal Genotyping Assay with Potential for Noninvasive Prenatal Diagnosis of Hereditary Hearing Loss. Prenat. Diagn. 2016, 36, 1233–1241. [Google Scholar] [CrossRef]
- Dan, S.; Yuan, Y.; Wang, Y.; Chen, C.; Gao, C.; Yu, S.; Liu, Y.; Song, W.; Zhu, H.; Yang, L.; et al. Non-Invasive Prenatal Diagnosis of Lethal Skeletal Dysplasia by Targeted Capture Sequencing of Maternal Plasma. PLoS ONE 2016, 11, e0159355. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Bijarnia-Mahay, S.; Saxena, R.; Kohli, S.; Dua-Puri, R.; Verma, J.; Thomas, E.; Shigematsu, Y.; Yamaguchi, S.; Deb, R.; et al. Identification of Mutations, Genotype-Phenotype Correlation and Prenatal Diagnosis of Maple Syrup Urine Disease in Indian Patients. Eur. J. Med. Genet. 2015, 58, 471–478. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.; Ge, H.-J.; Xiao, B.; Zhang, Y.-Y.; Ying, X.-M.; Pan, X.-Y.; Wang, L.; Xie, W.-W.; Ni, L.; et al. Haplotype-Based Approach for Noninvasive Prenatal Tests of Duchenne Muscular Dystrophy Using Cell-Free Fetal DNA in Maternal Plasma. Genet. Med. 2015, 17, 889–896. [Google Scholar] [CrossRef] [Green Version]
- Lv, W.; Wei, X.; Guo, R.; Liu, Q.; Zheng, Y.; Chang, J.; Bai, T.; Li, H.; Zhang, J.; Song, Z.; et al. Noninvasive Prenatal Testing for Wilson Disease by Use of Circulating Single-Molecule Amplification and Resequencing Technology (CSMART). Clin. Chem. 2015, 61, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Ge, H.; Li, X.; Jiang, T.; Chen, F.; Zhang, Y.; Hu, P.; Chen, S.; Zhang, J.; Ji, X.; et al. Haplotype-Based Approach for Noninvasive Prenatal Diagnosis of Congenital Adrenal Hyperplasia by Maternal Plasma DNA Sequencing. Gene 2014, 544, 252–258. [Google Scholar] [CrossRef]
- New, M.I.; Tong, Y.K.; Yuen, T.; Jiang, P.; Pina, C.; Chan, K.C.A.; Khattab, A.; Liao, G.J.W.; Yau, M.; Kim, S.-M.; et al. Noninvasive Prenatal Diagnosis of Congenital Adrenal Hyperplasia Using Cell-Free Fetal DNA in Maternal Plasma. J. Clin. Endocrinol. Metab. 2014, 99, E1022–E1030. [Google Scholar] [CrossRef] [Green Version]
- You, Y.; Sun, Y.; Li, X.; Li, Y.; Wei, X.; Chen, F.; Ge, H.; Lan, Z.; Zhu, Q.; Tang, Y.; et al. Integration of Targeted Sequencing and NIPT into Clinical Practice in a Chinese Family with Maple Syrup Urine Disease. Genet. Med. 2014, 16, 594–600. [Google Scholar] [CrossRef]
- D’Souza, E.; Sawant, P.M.; Nadkarni, A.H.; Gorakshakar, A.; Ghosh, K.; Colah, R.B. Detection of Fetal Mutations Causing Hemoglobinopathies by Non-Invasive Prenatal Diagnosis from Maternal Plasma. J. Postgrad. Med. 2013, 59, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Tsui, N.B.Y.; Kadir, R.A.; Chan, K.C.A.; Chi, C.; Mellars, G.; Tuddenham, E.G.; Leung, T.Y.; Lau, T.K.; Chiu, R.W.K.; Lo, Y.M.D. Noninvasive Prenatal Diagnosis of Hemophilia by Microfluidics Digital PCR Analysis of Maternal Plasma DNA. Blood 2011, 117, 3684–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khattab, A.; Yuen, T.; Sun, L.; Yau, M.; Barhan, A.; Zaidi, M.; Lo, Y.M.D.; New, M.I. Noninvasive Prenatal Diagnosis of Congenital Adrenal Hyperplasia. Endocr. Dev. 2016, 30, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazmi, D.; Bailey, J.; Yau, M.; Abu-Amer, W.; Kumar, A.; Low, M.; Yuen, T. New Developments in Prenatal Diagnosis of Congenital Adrenal Hyperplasia. J. Steroid. Biochem. Mol. Biol. 2017, 165, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Gong, H.; Wen, Y. Nucleic Acid-Based Non-Invasive Prenatal Diagnosis of Genetic Skin Diseases: Are We Ready? Exp. Dermatol. 2013, 22, 392–395. [Google Scholar] [CrossRef]
- Akiyama, M.; Titeux, M.; Sakai, K.; McMillan, J.R.; Tonasso, L.; Calvas, P.; Jossic, F.; Hovnanian, A.; Shimizu, H. DNA-Based Prenatal Diagnosis of Harlequin Ichthyosis and Characterization of ABCA12 Mutation Consequences. J. Investig. Dermatol. 2007, 127, 568–573. [Google Scholar] [CrossRef] [Green Version]
- Meij, K.R.M.; Kooij, C.; Bekker, M.N.; Galjaard, R.H.; Henneman, L. Dutch NIPT Consortium Non-invasive Prenatal Test Uptake in Socioeconomically Disadvantaged Neighborhoods. Prenat. Diagn. 2021, 41, 1395–1400. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, L.; Gao, J.; Li, S.; Chang, C.; Chen, Y.; Fei, H.; Zhang, J.; Wang, Y.; Huang, H.; et al. Expanding the Scope of Non-Invasive Prenatal Testing to Detect Fetal Chromosomal Copy Number Variations. Front. Mol. Biosci. 2021, 8, 649169. [Google Scholar] [CrossRef]
- Ge, Y.; Li, J.; Zhuang, J.; Zhang, J.; Huang, Y.; Tan, M.; Li, W.; Chen, J.; Zhou, Y. Expanded Noninvasive Prenatal Testing for Fetal Aneuploidy and Copy Number Variations and Parental Willingness for Invasive Diagnosis in a Cohort of 18,516 Cases. BMC Med. Genom. 2021, 14, 106. [Google Scholar] [CrossRef]
- Liang, D.; Cram, D.S.; Tan, H.; Linpeng, S.; Liu, Y.; Sun, H.; Zhang, Y.; Tian, F.; Zhu, H.; Xu, M.; et al. Clinical Utility of Noninvasive Prenatal Screening for Expanded Chromosome Disease Syndromes. Genet. Med. 2019, 21, 1998–2006. [Google Scholar] [CrossRef]
- van der Meij, K.R.M.; Sistermans, E.A.; Macville, M.V.E.; Stevens, S.J.C.; Bax, C.J.; Bekker, M.N.; Bilardo, C.M.; Boon, E.M.J.; Boter, M.; Diderich, K.E.M.; et al. TRIDENT-2: National Implementation of Genome-Wide Non-Invasive Prenatal Testing as a First-Tier Screening Test in the Netherlands. Am. J. Hum. Genet. 2019, 105, 1091–1101. [Google Scholar] [CrossRef]
- Kaseniit, K.E.; Hogan, G.J.; D’Auria, K.M.; Haverty, C.; Muzzey, D. Strategies to Minimize False Positives and Interpret Novel Microdeletions Based on Maternal Copy-Number Variants in 87,000 Noninvasive Prenatal Screens. BMC Med. Genom. 2018, 11, 90. [Google Scholar] [CrossRef] [Green Version]
- Petersen, A.K.; Cheung, S.W.; Smith, J.L.; Bi, W.; Ward, P.A.; Peacock, S.; Braxton, A.; Van Den Veyver, I.B.; Breman, A.M. Positive Predictive Value Estimates for Cell-Free Noninvasive Prenatal Screening from Data of a Large Referral Genetic Diagnostic Laboratory. Am. J. Obstet Gynecol 2017, 217, 691.e1–691.e6. [Google Scholar] [CrossRef] [Green Version]
- Vogel, I.; Vestergaard, E.M.; Lildballe, D.L.; Christensen, R.; Hoseth, G.-E.; Petersen, A.C.; Bogaard, P.; Sørensen, A.N. Placental Mosaicism in the Era of Chromosomal Microarrays. Eur. J. Med. Genet. 2020, 63, 103778. [Google Scholar] [CrossRef]
- Eggenhuizen, G.M.; Go, A.; Koster, M.P.H.; Baart, E.B.; Galjaard, R.J. Confined Placental Mosaicism and the Association with Pregnancy Outcome and Fetal Growth: A Review of the Literature. Hum. Reprod. Update 2021, 27, 885–903. [Google Scholar] [CrossRef]
- Samura, O.; Okamoto, A. Causes of Aberrant Non-Invasive Prenatal Testing for Aneuploidy: A Systematic Review. Taiwanese J. Obstet. Gynecol. 2020, 59, 16–20. [Google Scholar] [CrossRef]
- Christiaens, L.; Chitty, L.S.; Langlois, S. Current Controversies in Prenatal Diagnosis: Expanded NIPT That Includes Conditions Other than Trisomies 13, 18, and 21 Should Be Offered. Prenat. Diag. 2021, 41, 1316–1323. [Google Scholar] [CrossRef]
- Toews, M.; Caulfield, T. Physician Liability and Non-Invasive Prenatal Testing. J. Obstet. Gynaecol. Can. 2014, 36, 907–914. [Google Scholar] [CrossRef]
- Zaami, S.; Orrico, A.; Signore, F.; Cavaliere, A.F.; Mazzi, M.; Marinelli, E. Ethical, Legal and Social Issues (ELSI) Associated with Non-Invasive Prenatal Testing: Reflections on the Evolution of Prenatal Diagnosis and Procreative Choices. Genes 2021, 12, 204. [Google Scholar] [CrossRef]
- Pioro, M.; Mykitiuk, R.; Nisker, J. Wrongful Birth Litigation and Prenatal Screening. CMAJ 2008, 179, 1027–1030. [Google Scholar] [CrossRef]
- Hassan, M.; Chitty, L.; Reardon, H. Wrongful Birth: Clinical Settings and Legal Implications. Semin. Fetal Neonatal. Med. 2014, 19, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Kater-Kuipers, A.; Bunnik, E.M.; de Beaufort, I.D.; Galjaard, R.J.H. Limits to the Scope of Non-Invasive Prenatal Testing (NIPT): An Analysis of the International Ethical Framework for Prenatal Screening and an Interview Study with Dutch Professionals. BMC Pregnancy Childbirth 2018, 18, 409. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Authors (et al.); Year | Number of Patients | Type of Study | 21 Trisomy Detection Rate (%) | 18 Trisomy Detection Rate (%) | 13 Trisomy Detection Rate (%) |
|---|---|---|---|---|---|
| Judah 2021 [4] | 1442 | Cohort and Review | 99 | 92,8 | 94,7 |
| Borth 2021 [5] | 13,607 | Cohort | 98.89 | 99.99 | 99.99 |
| Serapinas 2020 [6] | 850 | Cohort | 100 | 100 | / |
| Gil 2019 [7] | 997 | Cohort | 98.2 | 88.9 | 66.7 |
| Miltoft 2018 [8] | 597 | Cohort | 100 | 100 | 100 |
| Gil 2017 [9] | 661,473 | Review and meta-analysis | 99.7 | 97.9 | 99 |
| Taylor-Phillips 2016 [10] | Not specified | Review | 99.3 | 97.4 | 97.4 |
| Zhang 2015 [11] | 112,669 | Cohort | 99.17 | 98.24 | 100 |
| Porreco 2014 [12] | 3340 | Cohort | 100 | 92.3 | 87.5 |
| Lau 2014 [13] | 1982 | Cohort | 100 | 100 | 100 |
| Stumm 2014 [14] | 485 | Cohort | 95.2 | 100 | 100 |
| Liang 2013 [15] | 435 | Cohort | 100 | 100 | 100 |
| Median detection rate | 99.50 | 99.12 | 99.99 | ||
| Authors (et al.); Year | Patients | Pathologies | Genes |
|---|---|---|---|
| Van der Meij 2021 [47] | 15,562 | Not specified | Not specified |
| Songchan 2021 [48] | 11,903 | Not specified | Alterations in the number and/or fractions of chromosomes 1, 2, 3, 5, 9, 13, 16, 18 |
| Yunsheng 2021 [49] | 18,516 | Not specified | Alterations in the number and/or fractions of chromosomes 1, 4, 9, 11, 12, 13, 14, 15, 20 |
| Liang 2019 [50] | 94,085 | Aneuploidies, DiGeorge, 22q11.22 microduplication, PW/Angelman, Cri du Chat and other microdeletion/duplication syndromes | Alterations in the number and/or fractions of chromosomes 22, 21, 18, 15, 13, 5. |
| Van der Meij 2019 [51] | 73,239 | PW and other non-specified conditions | Alterations in the number and/or fractions of chromosomes 9, 12, 15 and others |
| Kaseniit 2018 [52] | 87,255 | Not specified | Alterations in the number and/or fractions of chromosomes 1, 4, 5, 13, 15, 22 |
| Petersen 2017 [53] | 712 | Aneuploidies, DiGeorge, 22q11.22 microduplication, PW/Angelman, Cri du Chat and other microdeletion/duplication syndromes | Alterations in the number and/or fractions of chromosomes 22, 21, 18, 15, 13, 5 |
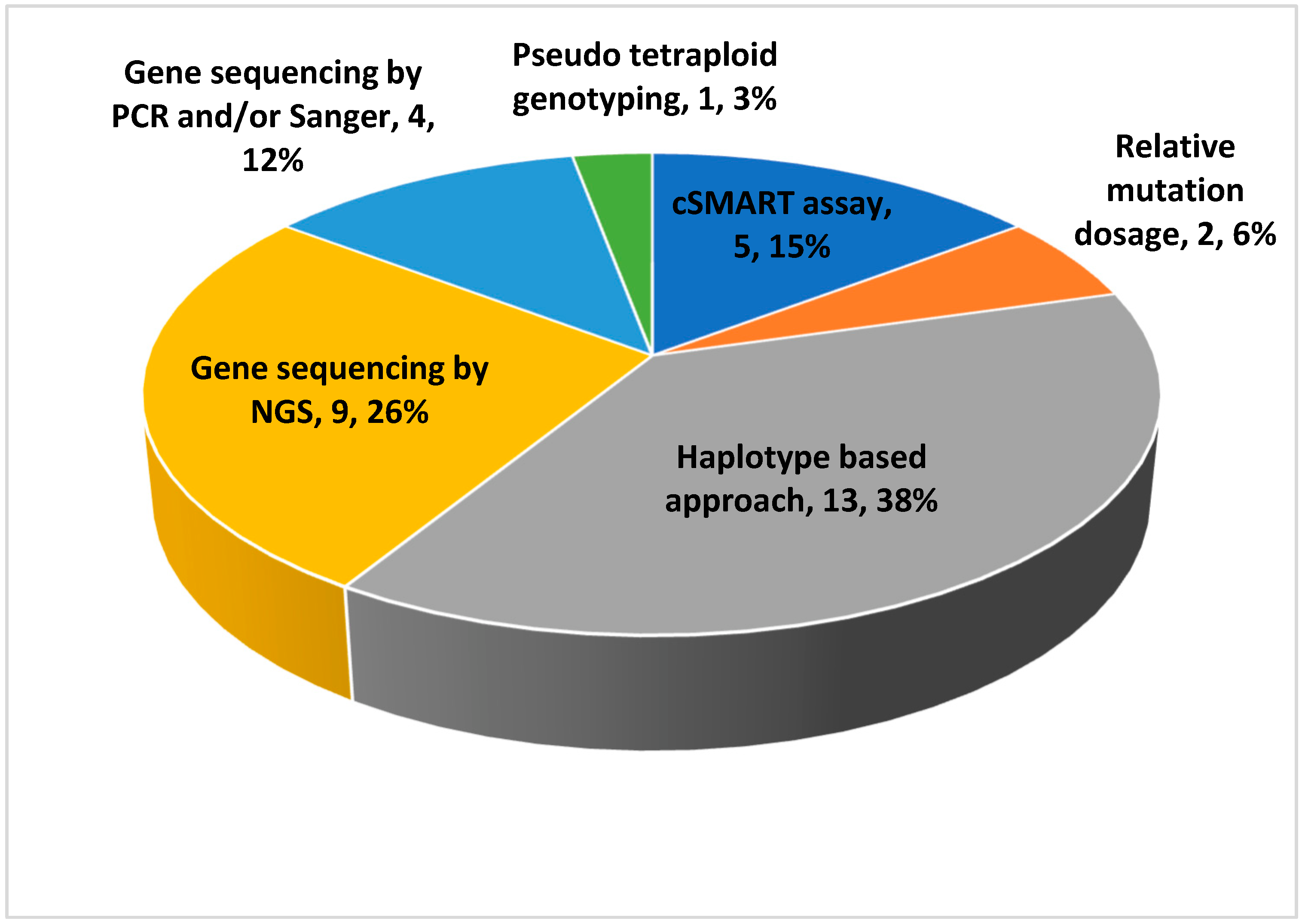
| Authors (et al.) | Year of Publication | Samples | Pathologies | Genes | Hereditary Transmission |
|---|---|---|---|---|---|
| Lv [17] | 2022 | 29 | Methylmalonic aciduria cblC type | MMACHC | AR |
| Zhao [18] | 2021 | 5 | Duchenne muscular dystrophy | DMD | X-Linked |
| Chen [2] | 2021 | 40 | Methylmalonic acidemia/aciduria, phenylketonuria, alfa/beta-thalassemia, ARPKD, DFNB1A | MMACHC, PAH, HBA, HBB, PKHD1, GJB2 | AR |
| Lv [19] | 2021 | 102 | Beta-thalassemia | HBB | AR |
| Kong [20] | 2021 | 21 | Duchenne muscular dystrophy | DMD | X-Linked |
| Wang [21] | 2021 | 59 | Skeletal dysplasia | FGFR2, FGFR3, COL1A1, COL1A2 and COL2A1 | AD |
| Morshneva [22] | 2021 | 645 | Mitochondrial disorders | mtDNA variants | Variable |
| De Falco [23] | 2021 | 1 | 46 XY disorders of sex development | HSD17B3 | Variable |
| Yang [24] | 2020 | 8 | Alpha and beta-thalassemia | HBA and HBB | AR |
| Jang [25] | 2018 | 5 | Duchenne muscular dystrophy | DMD | X-Linked |
| Yin [26] | 2018 | 1 | Osteogenesis imperfecta | COL1A1 | AD |
| Bijarnia-Mahay [27] | 2018 | 123 | Urea cycle disorders | ASS1, ASL, OTC, ARG1, CPS1, NAGS, SLC25A13, SLC7A7 | X-Linked (OTC) and AR |
| Ye [28] | 2018 | 13 | Hyperphenylalaninemia | PAH | AR |
| Parks [29] | 2017 | 6 | Spinal muscular atrophy | SMN1 | AR |
| Han [30] | 2017 | 80 | Non-syndromic hearing loss | GJB2 and SLC26A4 | AR |
| Vermeulen [31] | 2017 | 18 | Cystic fibrosis, congenital adrenal hyperplasia and beta-thalassemia | CFTR, CYP21A2, and HBB | AR |
| Ma [32] | 2017 | 14 | Congenital adrenal hyperplasia | CYP21A2 | AR |
| Chen [33] | 2016 | 25 | Non-syndromic hearing loss | GJB2, GJB3 and SLC26A4 | AR |
| Dan [34] | 2016 | 3 | Thanatophoric dysplasia, osteogenesis imperfecta type II, and achondroplasia | FGFR3, COL1A1 and COL2A2 | AD |
| Gupta [35] | 2015 | 24 | Maple syrup urine disease | BCKDHA, BCKDHB, DBT | AR |
| Xu [36] | 2015 | 8 | Duchenne muscular dystrophy | DMD | X-Linked |
| Lv [37] | 2015 | 4 | Wilson disease | ATP7B | AR |
| Ma [38] | 2014 | 1 | Congenital adrenal hyperplasia | CYP21A2 | AR |
| New [39] | 2014 | 14 | Congenital adrenal hyperplasia | CYP21A2 | AR |
| You [40] | 2014 | 1 | Maple syrup urine disease | BCKDHA | AR |
| D’souza [41] | 2013 | 30 | Beta-thalassemia | HBB | AR |
| Tsui [42] | 2011 | 12 | Hemophilia A and B | F8, F9 | X-Linked |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gullo, G.; Scaglione, M.; Buzzaccarini, G.; Laganà, A.S.; Basile, G.; Chiantera, V.; Cucinella, G.; Zaami, S. Cell-Free Fetal DNA and Non-Invasive Prenatal Diagnosis of Chromosomopathies and Pediatric Monogenic Diseases: A Critical Appraisal and Medicolegal Remarks. J. Pers. Med. 2023, 13, 1. https://doi.org/10.3390/jpm13010001
Gullo G, Scaglione M, Buzzaccarini G, Laganà AS, Basile G, Chiantera V, Cucinella G, Zaami S. Cell-Free Fetal DNA and Non-Invasive Prenatal Diagnosis of Chromosomopathies and Pediatric Monogenic Diseases: A Critical Appraisal and Medicolegal Remarks. Journal of Personalized Medicine. 2023; 13(1):1. https://doi.org/10.3390/jpm13010001
Chicago/Turabian StyleGullo, Giuseppe, Marco Scaglione, Giovanni Buzzaccarini, Antonio Simone Laganà, Giuseppe Basile, Vito Chiantera, Gaspare Cucinella, and Simona Zaami. 2023. "Cell-Free Fetal DNA and Non-Invasive Prenatal Diagnosis of Chromosomopathies and Pediatric Monogenic Diseases: A Critical Appraisal and Medicolegal Remarks" Journal of Personalized Medicine 13, no. 1: 1. https://doi.org/10.3390/jpm13010001