




Cutaneous Manifestations of Rheumatoid Arthritis: Diagnosis and Treatment

 ,
,  ,
,
Abstract
1. Introduction
2. Methods
3. Results
3.1. Nodules
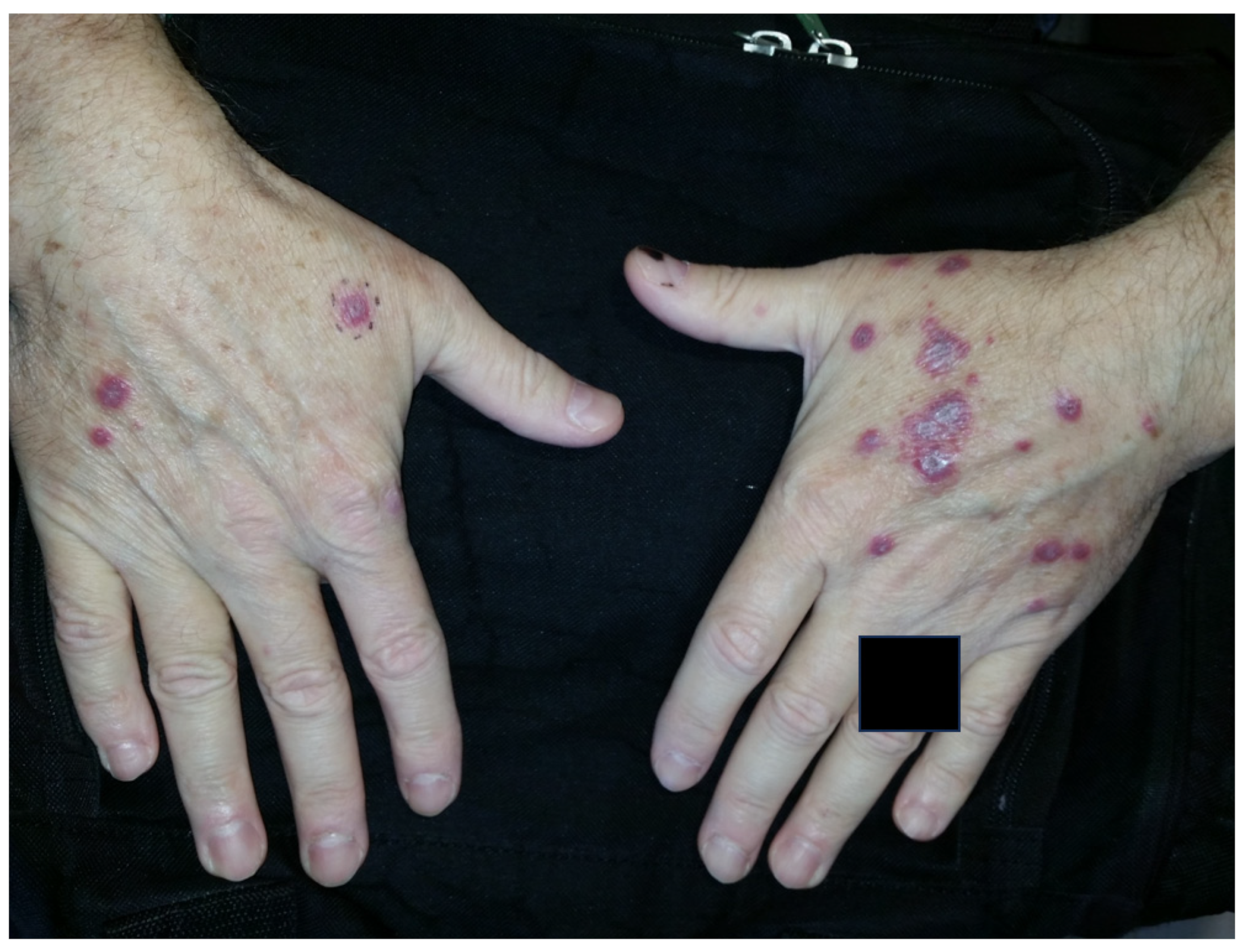
3.1.1. Rheumatoid Nodules
3.1.2. Rheumatoid Nodulosis
3.1.3. Accelerated Rheumatoid Nodulosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Etiology | Histology | Affected Areas | Treatment |
|---|---|---|---|---|
| Rheumatoid nodules | -Positive RF -Smoking | -Palisading macrophages -Granulation tissue | -Hands, fingers, elbows, feet | -SSZ, TCZ, RIX, colchicine, DPA -Corticosteroid injections -Surgery |
| Accelerated rheumatoid nodulosis | -MTX use -HLA-DRB1*0401 | -Palisading macrophages -Granulation tissue | -Hand, feet, ear | -Stop triggering drug -HCQ, DPA, SSZ, colchicine |
| Rheumatoid nodulosis | -No erosive arthritis -Negative RF | -Palisading macrophages -Granulation tissue | -Pretibial areas, feet, scalp, malleoli | -Prednisone, NSAIDs -Topical TAC, HCQ, DPA, PN -Surgery |
3.2. Neutrophilic Dermatoses
3.2.1. Sweet Syndrome
3.2.2. Pyoderma Gangrenosum
3.2.3. Rheumatoid Neutrophilic Dermatitis
3.2.4. Palisaded Neutrophilic Granulomatous Dermatitis and Interstitial Granulomatous Dermatitis
| Diagnosis | Clinical Characteristics | Histological Characteristics | Differential Diagnosis | Treatment |
|---|---|---|---|---|
| Sweet Syndrome | Acute-onset tender plaques or nodules, fever, arthralgia, ophthalmologic manifestations, headaches, oral or genital lesions | -Diffuse, dense neutrophilic infiltrate in the reticular dermis with interstitial leukocytoclastic nuclear debris -Epidermis often spared, but spongiosis and subcorneal pustules may be present | -Clinically: infection -Histologically: bowel bypass syndrome, erythema elevatum diutinum, granuloma faciale, halogenoderma, leukemia cutis, leukocytoclastic vasculitis, lobular neutrophilic panniculitides, neutrophilic eccrine hidradenitis, pyoderma gangrenosum, rheumatoid neutrophilic dermatitis | -First-line: systemic or topical corticosteroids, potassium iodide, colchicine -Second-line: indomethacin, clofazimine, cyclosporine, dapsone -Reported success in refractory cases: rituximab, anti-TNF-α agents, IL1 receptor antagonists, and granulocyte and monocyte adsorption apheresis |
| Pyoderma Gangrenosum | -Painful skin ulcers with undermined borders and peripheral erythema -Lesions can rapidly progress to blistered or necrotic ulcers | Intense neutrophilic infiltrate with neutrophilic pustules and abscess formation | Clinically: other causes of cutaneous ulceration including infections, malignancy, vascular ulceration, and systemic conditions such as systemic lupus erythematosus or Wegner’s granulomatosis -No definitive histopathological criteria | -Treatment includes anti-inflammatory therapies and proper wound care -First-line: fast-acting immunosuppressive drugs including topical or systemic corticosteroids and/or cyclosporine -Second-line: infliximab, other biologic TNF-α inhibitors, dapsone, minocycline -Refractory disease: IV immune globulin and alkylating agents |
| Rheumatoid neutrophilic dermatitis | Erythematous papules, nodules, plaques, and urticaria-like lesions | Dermal neutrophilic infiltrate with variable degree of leukocytoclasis in the absence of vasculitis | -Clinically: Sweet syndrome, pyoderma gangrenosum, Behcet disease, bowel bypass syndrome, rheumatoid nodules -Histologically: Sweet syndrome (difficult to distinguish based on similar histopathologic presentations) | -Topical or systemic corticosteroids, dapsone or colchicine -Efficacy demonstrated with cyclophosphamide and hydroxychloroquine |
| PNGD and IGD | -PNGD: symmetric smooth, umbilicated, or crusted papules, often on the elbows and extremities with skin to erythematous color -IGD: shared clinical features with PNGD, with locations favoring the lateral trunk and skin folds | -IGD: lympho-histiocytic infiltrate -PNGD: Greater neutrophil infiltrate | -PNGD, histologically: leukocytoclastic vasculitis, Sweet syndrome, neutrophilic urticaria | -Management of underlying condition -Noted clinical improvement with topical or oral corticosteroids, oral dapsone, and hydroxychloroquine |
3.3. Vasculitis and Vasculopathy
3.3.1. Felty Syndrome
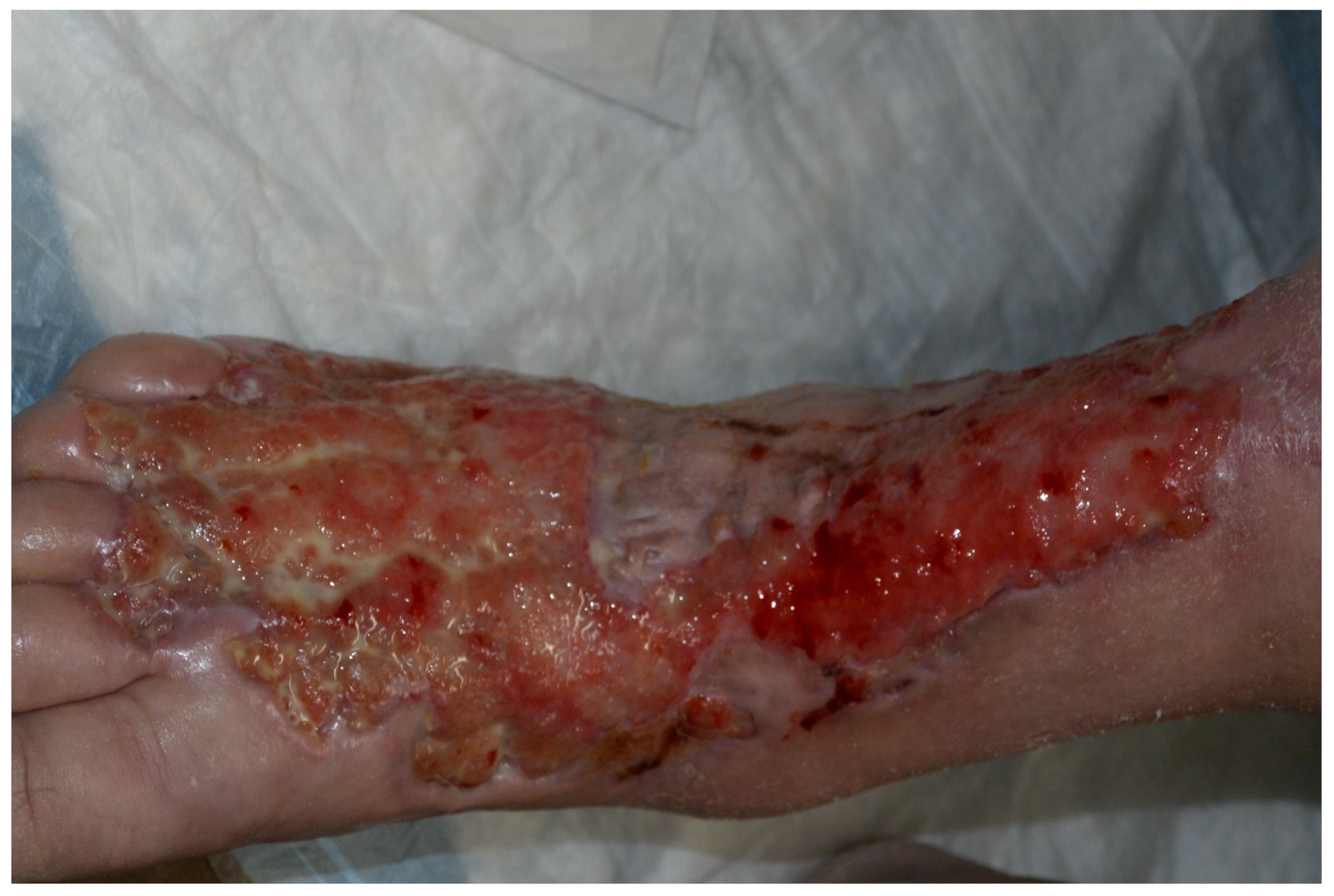
3.3.2. Rheumatoid Vasculitis
| Diagnosis | Characteristics | Clinical Findings | Differential Diagnosis | Treatment |
|---|---|---|---|---|
| Felty Syndrome | Severe RA features (erosive joint disease and deformity), Neutropenia, splenomegaly, vasculitis, necrotizing skin lesions, portal HTN, increased risk of malignancies, anemia of chronic disease, myeloid hyperplasia in bone marrow | Severe joint pain, swelling and deformities (esp. small joints of hands and wrists), fatigue, respiratory tract and skin bacterial infections, splenomegaly, variceal bleeding | SLE, large granular lymphocytic leukemia, other hematologic malignancies, drug reactions, amyloidosis, sarcoidosis, HIV, EBV | -MTX, glucocorticoids -Targeted immunotherapies: rituximab, tocilizumab -IVIG adjuvant therapy -Splenectomy |
| Rheumatoid Vasculitis | Severe RA features, cutaneous vasculitis, medium-sized artery necrotizing vasculitis, vasculitic peripheral neuropathy, ocular disease, cardiac disease, low serum complement C3 | RA symptoms, deep leg ulcers, palpable purpura, rash, sores around nails, constitutional symptoms (fever, fatigue, weight loss), sensory neuropathy, visual disturbances | -Non-vasculitic RA cutaneous syndromes: Sweet syndrome, pyoderma gangrenosum -Other vasculitides: PAN, ANCA-associated, cryoglobulinemic, paraneoplastic -Vasculitis mimics: Infection, malignancy, endocarditis, thromboembolic diseases | -Corticosteroids and immunosuppressive therapy -Targeted immunotherapies: rituximab, infliximab, etanercept, tocilizumab, peficitinib |
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Conforti, A.; Di Cola, I.; Pavlych, V.; Ruscitti, P.; Berardicurti, O.; Ursini, F.; Giacomelli, R.; Cipriani, P. Beyond the joints, the extra-articular manifestations in rheumatoid arthritis. Autoimmun. Rev. 2021, 20, 102735. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Atzeni, F.; Balsa, A.; Gossec, L.; Müller-Ladner, U.; Pope, J. The Key Comorbidities in Patients with Rheumatoid Arthritis: A Narrative Review. J. Clin. Med. 2021, 10, 509. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Yang, H.-Y.; Luo, S.-F.; Lai, J.-H. From Rheumatoid Factor to Anti-Citrullinated Protein Antibodies and Anti-Carbamylated Protein Antibodies for Diagnosis and Prognosis Prediction in Patients with Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 686. [Google Scholar] [CrossRef] [PubMed]
- Yap, H.-Y.; Tee, S.Z.-Y.; Wong, M.M.-T.; Chow, S.-K.; Peh, S.-C.; Teow, S.-Y. Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development. Cells 2018, 7, 161. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Deane, K.D.; Demoruelle, M.K.; Kelmenson, L.B.; Kuhn, K.A.; Norris, J.M.; Holers, V.M. Genetic and environmental risk factors for rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Maranini, B.; Bortoluzzi, A.; Silvagni, E.; Govoni, M. Focus on Sex and Gender: What We Need to Know in the Management of Rheumatoid Arthritis. J. Pers. Med. 2022, 12, 499. [Google Scholar] [CrossRef]
- Bullock, J.; Rizvi, S.A.A.; Saleh, A.M.; Ahmed, S.S.; Do, D.P.; Ansari, R.A.; Ahmed, J. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med. Princ. Pract. 2018, 27, 501–507. [Google Scholar] [CrossRef]
- Fraenkel, L.; Bathon, J.M.; England, B.R.; St Clair, E.W.; Arayssi, T.; Carandang, K.; Deane, K.D.; Genovese, M.; Huston, K.K.; Kerr, G.; et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2021, 73, 1108–1123. [Google Scholar] [CrossRef]
- Kaiser, M.-J.; Bozonnat, M.-C.; Jorgensen, C.; Daurès, J.-P.; Sany, J. Effect of etanercept on tenosynovitis and nodules in rheumatoid arthritis. Arthritis Rheum. 2002, 46, 559–560. [Google Scholar] [CrossRef]
- Khoury, T.; Ilan, Y. Introducing Patterns of Variability for Overcoming Compensatory Adaptation of the Immune System to Immunomodulatory Agents: A Novel Method for Improving Clinical Response to Anti-TNF Therapies. Front. Immunol. 2019, 10, 2726. [Google Scholar] [CrossRef] [PubMed]
- Jorizzo, J.L.; Daniels, J.C. Dermatologic conditions reported in patients with rheumatoid arthritis. J. Am. Acad. Dermatol. 1983, 8, 439–457. [Google Scholar] [CrossRef]
- Tilstra, J.S.; Lienesch, D.W. Rheumatoid Nodules. Dermatol. Clin. 2015, 33, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.G.; Hogg, N.; Highton, J.; Hessian, P.A.; Denholm, I. Macrophage migration and maturation within rheumatoid nodules. Arthritis Rheum. 1987, 30, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Veys, E.M.; De Keyser, F. Rheumatoid nodules: Differential diagnosis and immunohistological findings. Ann. Rheum. Dis. 1993, 52, 625–626. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Griffith, S.M.; Shadforth, M.F.; Fisher, J.; Dawes, P.T.; Poulton, K.V.; Thomson, W.; Ollier, W.E.R.; Mattey, D.L. Can gold therapy be used more safely in rheumatoid arthritis? Adverse drug reactions are more likely in patients with nodular disease, independent of HLA-DR3 status. J. Rheumatol. 2004, 31, 1903–1905. [Google Scholar] [PubMed]
- Langevitz, P.; Maguire, L.; Urowitz, M. Accelerated nodulosis during azathioprine therapy. Arthritis Rheum. 1991, 34, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Gómez, M.T.B.; Polo, A.M.; Romero, A.M.; Gutiérrez, J.V.; García, G.M. Rheumatoid nodulosis: Report of two cases. J. Eur. Acad. Dermatol. Venereol. 2003, 17, 695–698. [Google Scholar] [CrossRef]
- Gotsman, I.; Goral, A.; Nusair, S. Secondary spontaneous pneumothorax in a patient with pulmonary rheumatoid nodules during treatment with methotrexate. Rheumatology 2001, 40, 350–351. [Google Scholar] [CrossRef][Green Version]
- Toussirot, E.; Berthelot, J.M.; Pertuiset, E.; Bouvard, B.; Gaudin, P.; Wendling, D.; le Noach, J.; Lohse, A.; Lecuyer, E.; Cri, L. Pulmonary nodulosis and aseptic granulomatous lung disease occurring in patients with rheumatoid arthritis receiving tumor necrosis factor-α-blocking agent: A case series. J. Rheumatol. 2009, 36, 2421–2427. [Google Scholar] [CrossRef]
- Ariza-Hutchinson, A.; Patel, R.A.; Emil, N.S.; Muruganandam, M.; Nunez, S.E.; McElwee, M.K.; O’Sullivan, F.X.; Fields, R.A.; Hayward, W.A.; Haseler, L.J.; et al. Outcomes and Complications of Corticosteroid Injection of Rheumatoid Nodules. J. Clin. Aesthet. Dermatol. 2022, 15, 47–51. [Google Scholar] [PubMed]
- McGrath, M.H.; Fleischer, A. The subcutaneous rheumatoid nodule. Hand Clin. 1989, 5, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.W. Use of Cellular and Biocellular Therapy in Musculoskeletal Pain, Dysfunction, Degenerative or Inflammatory Disease; Clinicaltrials.gov. 2020. Available online: https://clinicaltrials.gov/study/NCT03090672 (accessed on 16 August 2023).
- Maldonado, I.; Eid, H.; Rodriguez, G.R.; Rillo, O.L.; Barcat, J.A.; Reginato, A.J. Rheumatoid nodulosis: Is it a different subset of rheumatoid arthritis? J. Clin. Rheumatol. 2003, 9, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Roux, F.; Wattiaux, M.-J.; Hayem, G.; Palazzo, E.; Kahn, M.-F.; Meyer, O. Rheumatoid nodulosis. Two cases with destructive polyarthritis after 20 years. Jt. Bone Spine 2006, 73, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Goñi, M.A.; Scheines, E.J.; Paira, S.O.; Barceló, H.A.; Maldonado Cocco, J.A. Rheumatoid nodulosis: A puzzling variant of rheumatoid arthritis. Clin. Rheumatol. 1992, 11, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Chua-Aguilera, C.J.; Möller, B.; Yawalkar, N. Skin Manifestations of Rheumatoid Arthritis, Juvenile Idiopathic Arthritis, and Spondyloarthritides. Clin. Rev. Allergy Immunol. 2017, 53, 371–393. [Google Scholar] [CrossRef] [PubMed]
- Wisnieski, J.J.; Askari, A.D. Rheumatoid nodulosis. A relatively benign rheumatoid variant. Arch. Intern. Med. 1981, 141, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.M.; Hadler, N.M.; Sams, W.M.; Wilson, J.; Snyderman, R. Rheumatoid nodulosis. Sporadic and familial diseases. J. Rheumatol. 1979, 6, 286–292. [Google Scholar] [PubMed]
- Baan, H.; Haagsma, C.J.; van de Laar, M.A.F.J. Corticosteroid injections reduce size of rheumatoid nodules. Clin. Rheumatol. 2006, 25, 21–23. [Google Scholar] [CrossRef]
- Garrido-Ríos, A.; Sánchez-Velicia, L.; Sanz-Muñoz, C.; Alvarez-Garrido, H.; Miranda-Romero, A. Rheumatoid nodulosis: Successful response to topical tacrolimus. Clin. Rheumatol. 2009, 28, 1341–1342. [Google Scholar] [CrossRef]
- McCarty, D.J. Complete reversal of rheumatoid nodulosis. J. Rheumatol. 1991, 18, 736–737. [Google Scholar] [PubMed]
- Kai, Y.; Anzai, S.; Shibuya, H.; Fujiwara, S.; Takayasu, S.; Asada, Y.; Terashi, H. A case of rheumatoid nodulosis successfully treated with surgery. J. Dermatol. 2004, 31, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, A.; McGrath, M.H. Rheumatoid nodulosis of the hand. J. Hand Surg. Am. 1984, 9, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.S.; Arnett, F.C.; Smith, C.A.; Ahn, C.; Reveille, J.D. The HLA-DRB1*0401 allele and the development of methotrexate-induced accelerated rheumatoid nodulosis: A follow-up study of 79 Caucasian patients with rheumatoid arthritis. Medicine 2001, 80, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Patatanian, E.; Thompson, D.F. A review of methotrexate-induced accelerated nodulosis. Pharmacotherapy 2002, 22, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Berkun, Y.; Abou Atta, I.; Rubinow, A.; Orbach, H.; Levartovsky, D.; Aamar, S.; Arbel, O.; Dresner-Pollak, R.; Friedman, G.; Ben-Yehuda, A. 2756GG genotype of methionine synthase reductase gene is more prevalent in rheumatoid arthritis patients treated with methotrexate and is associated with methotrexate-induced nodulosis. J. Rheumatol. 2007, 34, 1664–1669. [Google Scholar] [PubMed]
- Kerstens, P.J.; Boerbooms, A.M.; Jeurissen, M.E.; Fast, J.H.; Assmann, K.J.; van de Putte, L.B. Accelerated nodulosis during low dose methotrexate therapy for rheumatoid arthritis. An analysis of ten cases. J. Rheumatol. 1992, 19, 867–871. [Google Scholar] [PubMed]
- Palmeiro, A.G.; Lourenço, M.H.; Miroux-Catarino, A.; Crispim, I.; Branco, J.C.; Viana, I. Drug-induced accelerated nodulosis: Review of the literature. Int. J. Dermatol. 2023, 62, 432–440. [Google Scholar] [CrossRef]
- Merrill, J.T.; Shen, C.; Schreibman, D.; Coffey, D.; Zakharenko, O.; Fisher, R.; Lahita, R.G.; Salmon, J.; Cronstein, B.N. Adenosine A1 receptor promotion of multinucleated giant cell formation by human monocytes. A mechanism for methotrexate-induced nodulosis in rheumatoid arthritis. Arthritis Rheum. 1997, 40, 1308–1315. [Google Scholar] [CrossRef]
- Thompson, R.N. Is the appearance of rheumatoid nodules in a rheumatoid patient well controlled on methotrexate an indication to stop the drug? Br. J. Rheumatol. 1992, 31, 86. [Google Scholar] [CrossRef]
- Callen, J.P. Neutrophilic dermatoses. Dermatol. Clin. 2002, 20, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, P.; Goyal, A.; Hearth Holmes, M.P. Sweet Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK431050/ (accessed on 19 August 2023).
- Cohen, P.R. Sweet’s syndrome—A comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J. Rare Dis. 2007, 2, 34. [Google Scholar] [CrossRef]
- Seminario-Vidal, L.; Guerrero, C.; Sami, N. Refractory Sweet’s syndrome successfully treated with rituximab. JAAD Case Rep. 2015, 1, 123–125. [Google Scholar] [CrossRef][Green Version]
- Foster, E.N.; Nguyen, K.K.; Sheikh, R.A.; Prindiville, T.P. Crohn’s disease associated with Sweet’s syndrome and Sjögren’s syndrome treated with infliximab. Clin. Dev. Immunol. 2005, 12, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Kluger, N.; Gil-Bistes, D.; Guillot, B.; Bessis, D. Efficacy of anti-interleukin-1 receptor antagonist anakinra (Kineret®) in a case of refractory Sweet’s syndrome. Dermatology 2011, 222, 123–127. [Google Scholar] [CrossRef]
- Delluc, A.; Limal, N.; Puéchal, X.; Francès, C.; Piette, J.C.; Cacoub, P. Efficacy of anakinra, an IL1 receptor antagonist, in refractory Sweet syndrome. Ann. Rheum. Dis. 2008, 67, 278–279. [Google Scholar] [CrossRef] [PubMed]
- Fujii, A.; Mizutani, Y.; Hattori, Y.; Takahashi, T.; Ohnishi, H.; Yoshida, S.; Seishima, M. Sweet’s Syndrome Successfully Treated with Granulocyte and Monocyte Adsorption Apheresis. Case Rep. Dermatol. 2017, 9, 13–18. [Google Scholar] [CrossRef]
- Joshi, T.P.; Friske, S.K.; Hsiou, D.A.; Duvic, M. New Practical Aspects of Sweet Syndrome. Am. J. Clin. Dermatol. 2022, 23, 301–318. [Google Scholar] [CrossRef]
- Ravi, M.; Waters, M.; Trinidad, J.; Chung, C.G.; Kaffenberger, B.H. A retrospective cohort study of infection risk in hospitalized patients evaluated for Sweet syndrome. Arch. Dermatol. Res. 2022, 314, 967–973. [Google Scholar] [CrossRef]
- Maverakis, E.; Marzano, A.V.; Le, S.T.; Callen, J.P.; Brüggen, M.-C.; Guenova, E.; Dissemond, J.; Shinkai, K.; Langan, S.M. Pyoderma gangrenosum. Nat. Rev. Dis. Primers 2020, 6, 81. [Google Scholar] [CrossRef]
- George, C.; Deroide, F.; Rustin, M. Pyoderma gangrenosum—A guide to diagnosis and management. Clin. Med. 2019, 19, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Brooklyn, T.; Dunnill, G.; Probert, C. Diagnosis and treatment of pyoderma gangrenosum. BMJ 2006, 333, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Nusbaum, K.B.; Korman, A.M.; Tyler, K.H.; Kaffenberger, J.A.; Trinidad, J.C.; Dean, S.; Cataland, S.; Kaffenberger, B.H. In vitro diagnostics for the medical dermatologist. Part II: Hypercoagulability tests. J. Am. Acad. Dermatol. 2021, 85, 301–310. [Google Scholar] [CrossRef]
- Nusbaum, K.B.; Korman, A.M.; Tyler, K.; Kaffenberger, J.; Trinidad, J.; Kaffenberger, B.H. In vitro diagnostics for the medical dermatologist. Part I: Autoimmune tests. J. Am. Acad. Dermatol. 2021, 85, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Ravi, M.; Trinidad, J.; Spaccarelli, N.; Kaffenberger, B. The impact of comorbidity identification on outcomes in patients with pyoderma gangrenosum: A retrospective cohort study of previously hospitalized patients. J. Am. Acad. Dermatol. 2022, 87, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, A.D.; Thomas, K.S.; Craig, F.E.; Mitchell, E.; Greenlaw, N.; Norrie, J.; Mason, J.M.; Walton, S.; Johnston, G.A.; Williams, H.C.; et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: Results of the STOP GAP randomised controlled trial. BMJ 2015, 350, h2958. [Google Scholar] [CrossRef] [PubMed]
- Westerdahl, J.S.; Nusbaum, K.B.; Chung, C.G.; Kaffenberger, B.H.; Ortega-Loayza, A.G. Ustekinumab as adjuvant treatment for all pyoderma gangrenosum subtypes. J. Dermatol. Treat. 2022, 33, 2386–2390. [Google Scholar] [CrossRef]
- Azuine, M.A.; Kayal, J.J.; Bhide, S.V. Protective role of aqueous turmeric extract against mutagenicity of direct-acting carcinogens as well as benzo [α] pyrene-induced genotoxicity and carcinogenicity. J. Cancer Res. Clin. Oncol. 1992, 118, 447–452. [Google Scholar] [CrossRef]
- Yamasaki, K.; Yamanaka, K.; Zhao, Y.; Iwano, S.; Takei, K.; Suzuki, K.; Yamamoto, T. Adalimumab in Japanese patients with active ulcers of pyoderma gangrenosum: Final analysis of a 52-week phase 3 open-label study. J. Dermatol. 2022, 49, 479–487. [Google Scholar] [CrossRef]
- Rousset, L.; de Masson, A.; Begon, E.; Villani, A.; Battistella, M.; Rybojad, M.; Jachiet, M.; Bagot, M.; Bouaziz, J.-D.; Lepelletier, C. Tumor necrosis factor-α inhibitors for the treatment of pyoderma gangrenosum not associated with inflammatory bowel diseases: A multicenter retrospective study. J. Am. Acad. Dermatol. 2019, 80, 1141–1143. [Google Scholar] [CrossRef]
- Herberger, K.; Dissemond, J.; Brüggestrat, S.; Sorbe, C.; Augustin, M. Biologics and immunoglobulins in the treatment of pyoderma gangrenosum—Analysis of 52 patients. J. Dtsch. Dermatol. Ges. 2019, 17, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Lahood, N.; Mostaghimi, A. Intravenous immunoglobulin as adjunct therapy for refractory pyoderma gangrenosum: Systematic review of cases and case series. Br. J. Dermatol. 2018, 178, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Zaheer, M.; Balis, F.J. Baricitinib. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK572064/ (accessed on 19 August 2023).
- Loayza, A.O. Baricitinib in the Treatment of Adults with Pyoderma Gangrenosum (PG); Clinicaltrials.gov. 2023. Available online: https://clinicaltrials.gov/study/NCT04901325 (accessed on 16 August 2023).
- InflaRx GmbH Open Label Exploratory Phase 2a Trial to Investigate the Safety and Efficacy of IFX-1 in Treating Subjects with Pyoderma Gangrenosum (Optima); Clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/study/NCT03971643 (accessed on 16 August 2023).
- Kaffenberger, B.H. An Open-Label, Proof-of-Concept Study of Ixekizumab in the Treatment of Pyoderma Gangrenosum; Clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/study/NCT03137160 (accessed on 16 August 2023).
- IbnIdris Rodwan, A.A.; Mohammed, A.G.; Adam Essa, M.E.; Abdalla Babker, A.E.; Mohamed Abdelsatir, A.; Mohammed Elagib, E. Neutrophilic dermatoses in a seronegative rheumatoid arthritis patient: A case report. Clin. Case Rep. 2022, 10, e05249. [Google Scholar] [CrossRef] [PubMed]
- Bevin, A.A.; Steger, J.; Mannino, S. Rheumatoid neutrophilic dermatitis. Cutis 2006, 78, 133–136. [Google Scholar] [PubMed]
- Jost, M.; Schwarz, T.; Wehkamp, U.; Bohne, A.-S.; Drerup, K. Rheumatoid neutrophilic dermatosis under treatment with the interleukin-6-receptor-antagonist sarilumab in a patient with seropositive rheumatoid arthritis. J. Cutan. Pathol. 2023, 50, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Manriquez, J.; Giesen, L.; Puerto, C.D.; Gonzalez, S. Rheumatoid arthritis and pseudo-vesicular skin plaques: Rheumatoid neutrophilic dermatosis. An. Bras. Dermatol. 2016, 91, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Mashek, H.A.; Pham, C.T.; Helm, T.N.; Klaus, M. Rheumatoid neutrophilic dermatitis. Arch. Dermatol. 1997, 133, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Schadt, C.R.; Callen, J.P. Management of neutrophilic dermatoses. Dermatol. Ther. 2012, 25, 158–172. [Google Scholar] [CrossRef]
- Rosenbach, M.; English, J.C. Reactive Granulomatous Dermatitis: A Review of Palisaded Neutrophilic and Granulomatous Dermatitis, Interstitial Granulomatous Dermatitis, Interstitial Granulomatous Drug Reaction, and a Proposed Reclassification. Dermatol. Clin. 2015, 33, 373–387. [Google Scholar] [CrossRef]
- Tomasini, C.; Pippione, M. Interstitial granulomatous dermatitis with plaques. J. Am. Acad. Dermatol. 2002, 46, 892–899. [Google Scholar] [CrossRef]
- Coutinho, I.; Pereira, N.; Gouveia, M.; Cardoso, J.C.; Tellechea, O. Interstitial Granulomatous Dermatitis: A Clinicopathological Study. Am. J. Dermatopathol. 2015, 37, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Garijo, N.; Bielsa, I.; Mascaró, J.M.; Quer, A.; Idoate, M.A.; Paricio, J.J.; Iranzo, P.; España, A. Reactive granulomatous dermatitis as a histological pattern including manifestations of interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis: A study of 52 patients. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Kalen, J.E.; Shokeen, D.; Ramos-Caro, F.; Motaparthi, K. Palisaded neutrophilic granulomatous dermatitis: Spectrum of histologic findings in a single patient. JAAD Case Rep. 2017, 3, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Brecher, A. Palisaded neutrophilic and granulomatous dermatitis. Dermatol. Online J. 2003, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Maverakis, E.; Ramirez-Maverakis, D.; Fitzmaurice, S. Combination therapy with intralesional triamcinolone and oral dapsone for management of palisaded neutrophilic and granulomatous dermatitis. Dermatol. Online J. 2013, 19, 17. [Google Scholar] [CrossRef] [PubMed]
- Fett, N.; Kovarik, C.; Bennett, D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J. Am. Acad. Dermatol. 2011, 65, e92–e93. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, S.; Itin, P.H.; Hasler, P. Arthritis and interstitial granulomatous dermatitis (Ackerman syndrome) with pulmonary silicosis. Semin. Arthritis Rheum. 2003, 32, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Walling, H.W.; Swick, B.L. Interstitial granulomatous dermatitis associated with chronic inflammatory demyelinating polyneuropathy. Cutis 2012, 90, 30–32. [Google Scholar] [PubMed]
- Gerbing, E.K.; Metze, D.; Luger, T.A.; Ständer, S. Interstitial granulomatous dermatitis without arthritis: Successful therapy with hydroxychloroquine. J. Dtsch. Dermatol. Ges. 2003, 1, 137–141. [Google Scholar] [PubMed]
- Elsawi, R.; Brooks, S.G.; Georgakopolous, J.R.; Lansang, P.; Piguet, V.; Croitoru, D. The Utility of IL-17 Inhibitors in Neutrophilic Dermatoses: A Systematic Review. J. Cutan. Med. Surg. 2022, 26, 210–211. [Google Scholar] [CrossRef]
- Owlia, M.B.; Newman, K.; Akhtari, M. Felty’s Syndrome, Insights and Updates. Open Rheumatol. J. 2014, 8, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, J.; Jones, R.; Silberman, H.R.; Banks, P.M. Splenectomy for Felty’s syndrome. Clinicopathological study of 27 patients. Arch. Intern. Med. 1978, 138, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Lora, V.; Cerroni, L.; Cota, C. Skin manifestations of rheumatoid arthritis. G. Ital. Dermatol. Venereol. 2018, 153, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.; Grisanti, M.W.; Grisanti, J.M. Oral methotrexate in the treatment of Felty’s syndrome. J. Rheumatol. 1993, 20, 599–601. [Google Scholar] [PubMed]
- Patel, R.; Akhondi, H. Felty Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK546693/ (accessed on 19 August 2023).
- Li, R.; Wan, Q.; Chen, P.; Mao, S.; Wang, Q.; Li, X.; Yang, Y.; Dong, L. Tocilizumab treatment in Felty’s syndrome. Rheumatol. Int. 2020, 40, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Narváez, J.; Domingo-Domenech, E.; Gómez-Vaquero, C.; López-Vives, L.; Estrada, P.; Aparicio, M.; Martín-Esteve, I.; Nolla, J.M. Biological agents in the management of Felty’s syndrome: A systematic review. Semin. Arthritis Rheum. 2012, 41, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Balint, G.P.; Balint, P.V. Felty’s syndrome. Best Pract. Res. Clin. Rheumatol. 2004, 18, 631–645. [Google Scholar] [CrossRef] [PubMed]
- de Cerqueira, D.P.A.; Pedreira, A.L.S.; de Cerqueira, M.G.; Santiago, M.B. Biological therapy in rheumatoid vasculitis: A systematic review. Clin. Rheumatol. 2021, 40, 1717–1724. [Google Scholar] [CrossRef]
- Olivé, A.; Riveros, A.; Juárez, P.; Morales-Ivorra, I.; Holgado, S.; Narváez, J. Rheumatoid vasculitis: Study of 41 cases. Med. Clin. 2020, 155, 126–129. [Google Scholar] [CrossRef]
- Makol, A.; Matteson, E.L.; Warrington, K.J. Rheumatoid vasculitis: An update. Curr. Opin. Rheumatol. 2015, 27, 63–70. [Google Scholar] [CrossRef]
- Coffey, C.M.; Richter, M.D.; Crowson, C.S.; Koster, M.J.; Warrington, K.J.; Ytterberg, S.R.; Makol, A. Rituximab Therapy for Systemic Rheumatoid Vasculitis: Indications, Outcomes, and Adverse Events. J. Rheumatol. 2020, 47, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Oba, Y.; Sawa, N.; Ikuma, D.; Mizuno, H.; Inoue, N.; Sekine, A.; Hasegawa, E.; Yamanouchi, M.; Suwabe, T.; Yamaguchi, Y.; et al. Successful Peficitinib Monotherapy for the New-Onset Skin Manifestations of Rheumatoid Vasculitis After Long-Term Treatment with Tocilizumab for Rheumatoid Arthritis. Mod. Rheumatol. Case Rep. 2023, rxad025. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz, M.J.; Natarelli, N.; Wei, A.; Rechdan, M.; Botto, E.; Tran, J.T.; Forouzandeh, M.; Plaza, J.A.; Kaffenberger, B.H. Cutaneous Manifestations of Rheumatoid Arthritis: Diagnosis and Treatment. J. Pers. Med. 2023, 13, 1479. https://doi.org/10.3390/jpm13101479
Diaz MJ, Natarelli N, Wei A, Rechdan M, Botto E, Tran JT, Forouzandeh M, Plaza JA, Kaffenberger BH. Cutaneous Manifestations of Rheumatoid Arthritis: Diagnosis and Treatment. Journal of Personalized Medicine. 2023; 13(10):1479. https://doi.org/10.3390/jpm13101479
Chicago/Turabian StyleDiaz, Michael J., Nicole Natarelli, Aria Wei, Michaela Rechdan, Elizabeth Botto, Jasmine T. Tran, Mahtab Forouzandeh, Jose A. Plaza, and Benjamin H. Kaffenberger. 2023. "Cutaneous Manifestations of Rheumatoid Arthritis: Diagnosis and Treatment" Journal of Personalized Medicine 13, no. 10: 1479. https://doi.org/10.3390/jpm13101479
APA StyleDiaz, M. J., Natarelli, N., Wei, A., Rechdan, M., Botto, E., Tran, J. T., Forouzandeh, M., Plaza, J. A., & Kaffenberger, B. H. (2023). Cutaneous Manifestations of Rheumatoid Arthritis: Diagnosis and Treatment. Journal of Personalized Medicine, 13(10), 1479. https://doi.org/10.3390/jpm13101479