
Lymphangioleiomyomatosis with Tuberous Sclerosis Complex—A Case Study

 , ,
, , 
{kind=link}
Abstract
:1. Introduction
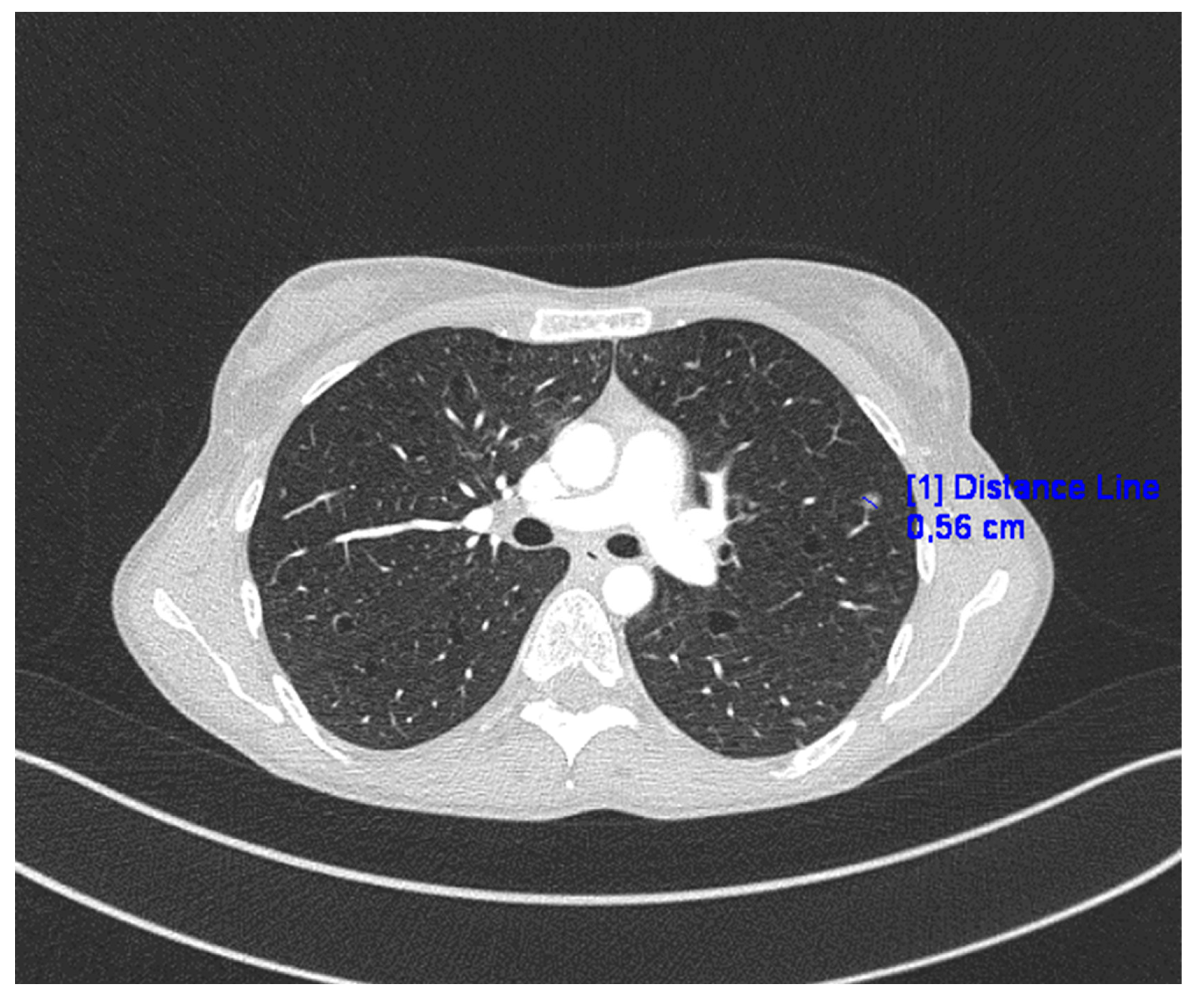
2. Case Presentation
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prizant, H.; Hammes, S. Minireview: Lymphangioleiomyomatosis (LAM): The “Other” Steroid-Sensitive Cancer. Endocrinology 2016, 157, 3374–3383. [Google Scholar] [CrossRef]
- McCarthy, C.; Gupta, N.; Johnson, S.R.; Yu, J.J.; McCormack, F.X. Lymphangioleiomyomatosis: Pathogenesis, clinical features, diagnosis, and management. Lancet Respir. Med. 2021, 9, 1313–1327. [Google Scholar] [CrossRef]
- Esposito, A.J.; Imani, J.; Shrestha, S.; Bagwe, S.; Lamattina, A.M.; Vivero, M.; Goldberg, H.J.; Rosas, I.O.; Henske, E.P.; El-Chemaly, S.Y. Lymphangioleiomyomatosis: Circulating Levels of FGF23 and pulmonary diffusion. J. Bras. Pneumol. 2023, 49, e20220356. [Google Scholar] [CrossRef]
- Shah, J.M.; Patel, J.T.; Shah, H.; Dadigiri, H.; Alla, A.; Cheriyath, P. The epidemiology and clinical features of Lymphangioleiomyomatosis (LAM): A descriptive study of 33 case reports. Cureus 2023, 15, e43513. [Google Scholar] [CrossRef]
- Ağaçkiran, Y.; Ertürk, A.; Yeşıller, F.I.; Hoca, N.T.; Ustün, L.N.; Capan, N. Pulmonary lymphangioleiomyomatosis: A rare case. Turk. Patoloji Derg. 2014, 30, 233–236. [Google Scholar]
- Xu, K.; Xu, W.; Liu, S.; Yu, J.; Tian, X.; Yang, Y.; Wang, S.; Zhang, W.; Feng, R.; Zhang, T. Lymphangioleiomyomatosis. Semin. Respir. Crit. Care Med. 2020, 41, 256–268. [Google Scholar] [CrossRef]
- Kundu, N.; Holz, M. Lymphangioleiomyomatosis: A metastatic lung diseas. Am. J. Physiol. Cell Physiol. 2023, 324, 320–326. [Google Scholar] [CrossRef]
- Nijmeh, J.; El-Chemaly, S.; Henske, E.P. Emerging biomarkers of lymphangioleiomyomatosis. Expert Rev. Respir. Med. 2018, 12, 95–102. [Google Scholar] [CrossRef]
- Cottin, V.; Archer, F.; Leroux, C.; Mornex, J.F.; Cordier, J.F. Milestones in lymphangioleiomyomatosis research. Eur. Respir. Rev. 2011, 20, 3–6. [Google Scholar] [CrossRef]
- McCormack, F.X. Lymphangioleiomyomatosis: A clinical update. Chest 2008, 133, 507–516. [Google Scholar] [CrossRef]
- Johnson, S.R.; Cordier, J.F.; Lazor, R.; Cottin, V.; Costabel, U.; Harari, S.; Reynaud-Gaubert, M.; Boehler, A.; Brauner, M.; Popper, H.; et al. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur. Respir. J. 2010, 35, 14–26. [Google Scholar] [CrossRef]
- McCormack, F.X.; Gupta, N.; Finlay, G.R.; Young, L.R.; Taveira-DaSilva, A.M.; Glasgow, C.G.; Steagall, W.K.; Johnson, S.R.; Sahn, S.A.; Ryu, J.H.; et al. Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines: Lymphangioleiomyomatosis Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2016, 194, 748–761. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, A.; Lynn, E.; Murphy, D.; Fabre, A.; McCarthy, C. Lymphangioleiomyomatosis: A clinical review. Breathe 2020, 16, 200007. [Google Scholar] [CrossRef]
- Gibbons, E.; Minor, B.; Hammes, S.R. Lymphangioleiomyomatosis: Where endocrinology, immunology and tumor biology meet. Endocr. Relat. Cancer 2023, 30, e230102. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Li, Y.; Li, H.; Yang, P.; Xing, X. Solitary extrapulmonary lymphangioleiomyomatosis of the liver: A case report and literature review. Exp. Ther. Med. 2016, 12, 1499–1502. [Google Scholar] [CrossRef]
- Song, D.H.; Choi, I.H.; Ha, S.Y.; Han, K.M.; Lee, J.J.; Hong, M.E.; Choi, Y.L.; Jang, K.T.; Song, S.Y.; Yi, C.A.; et al. Extrapulmonary lymphangioleiomyoma: Clinicopathological analysis of 4 cases. Korean J. Pathol. 2014, 48, 188–192. [Google Scholar] [CrossRef]
- Matsui, K.; Tatsuguchi, A.; Valencia, J.; Yu, Z.X.; Bechtle, J.; Beasley, M.B.; Avila, N.; Travis, W.D.; Moss, J.; Ferrans, V.J. Extrapulmonary lymphangioleiomyomatosis (LAM): Clinicopathologic features in 22 cases. Hum. Pathol. 2000, 31, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Park, M.I.; Suh, K.S. Lymphangiomyomatosis arising in the pelvic cavity: A case report. J. Korean Med. Sci. 2005, 20, 904–907. [Google Scholar] [CrossRef]
- Han, J.M.; Lee, K.H.; Kim, S.J.; Rhim, C.C.; Park, Y.H.; Kang, J.B.; Jeon, S.Y. A case of lymphangioleiomyomatosis originated in the pelvic cavity. J. Gynecol. Oncol. 2008, 19, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Ofori, K.; Fernandes, H.; Cummings, M.; Colby, T.; Saqia, A. Benign metastasizing leiomyoma presenting with miliary pattern and fatal outcome: Case report with molecular analysis & review of the literature. Respir. Med. Case Rep. 2019, 27, 100831. [Google Scholar] [CrossRef]
- Kingswood, J.C.; Bruzzi, P.; Curatolo, P.; De Vries, P.J.; Fladrowski, C.; Hertzberg, C.; Jansen, A.C.; Jozwiak, S.; Nabbout, R.; Sauter, M.; et al. TOSCA—First international registry to address knowledge gaps in the natural history and management of tuberous sclerosis complex. Orphanet. J. Rare Dis. 2014, 9, 182. [Google Scholar] [CrossRef]
- Chebib, N.; Archer, F.; Bobet-Erny, A.; Leroux, C.; Cottin, V. Dysregulation of the endothelin pathway in lymphangioleiomyomatosis with no direct effect on cell proliferation and migration. Sci. Rep. 2018, 8, 14698. [Google Scholar] [CrossRef] [PubMed]
- Cong, C.V.; Tuan Anh, T.; Ly, T.; Duc, N. Pulmonary lymphangioleiomyomatosis (LAM): A literature overview and case report. Radiol. Case Rep. 2022, 17, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Tümay, L.; Güner, O.; Zorluoğlu, A. An extrapulmonary manifestation of lymphangioleiomyomatosis: A rare case report. Int. J. Surg. Case Rep. 2017, 41, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, M.; Kołaczyk, K.; Chamier-Ciemińska, K.; Walecka, A.; Chosia, M.; Szydłowska, I.; Starczewski, A.; Grodzki, T.; Smereczyński, A. Pulmonary Benign Metastasizing Leiomyoma from the Uterine Leiomyoma: A Case Report. Pol. J. Radiol. 2015, 80, 107–110. [Google Scholar] [CrossRef]
- Moir, L.M. Lymphangioleiomyomatosis: Current understanding and potential treatments. Pharmacol. Ther. 2016, 158, 114–124. [Google Scholar] [CrossRef]
- Wahid, S.; Chiang, P.C.; Luo, H.L.; Huang, S.C.; Tsai, E.M.; Chiang, P.H. Pelvic lymphangioleiomyomatosis treated successfully with everolimus: Two case reports with literature review. Medicine 2017, 96, e4562. [Google Scholar] [CrossRef]
- Sathirareuangchai, S.; Shimizu, D.; Vierkoetter, K.R. Pulmonary Lymphangioleiomyomatosis: A Case Report and Literature Review. Hawaii J. Health Soc. Welf. 2020, 79, 224–229. [Google Scholar]
- Pfizer’s RAPAMUNE® (sirolimus) Becomes First FDA-Approved Treatment for Lymphangioleiomyomatosis (LAM), A Rare Progressive Lung Disease. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer_s_rapamune_sirolimus_becomes_first_fda_approved_treatment_for_lymphangioleiomyomatosis_lam_a_rare_progressive_lung_disease (accessed on 7 August 2023).
- Song, X.; Cai, H.; Yang, C.; Xue, X.; Wang, J.; Mo, Y.; Zhu, M.; Zhu, G.; Ye, L.; Jin, M. Possible Novel Therapeutic Targets in Lymphangioleiomyomatosis Treatment. Front. Med. 2020, 7, 554134. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marciniak, A.; Nawrocka-Rutkowska, J.; Brodowska, A.; Starczewski, A.; Szydłowska, I. Lymphangioleiomyomatosis with Tuberous Sclerosis Complex—A Case Study. J. Pers. Med. 2023, 13, 1598. https://doi.org/10.3390/jpm13111598
Marciniak A, Nawrocka-Rutkowska J, Brodowska A, Starczewski A, Szydłowska I. Lymphangioleiomyomatosis with Tuberous Sclerosis Complex—A Case Study. Journal of Personalized Medicine. 2023; 13(11):1598. https://doi.org/10.3390/jpm13111598
Chicago/Turabian StyleMarciniak, Aleksandra, Jolanta Nawrocka-Rutkowska, Agnieszka Brodowska, Andrzej Starczewski, and Iwona Szydłowska. 2023. "Lymphangioleiomyomatosis with Tuberous Sclerosis Complex—A Case Study" Journal of Personalized Medicine 13, no. 11: 1598. https://doi.org/10.3390/jpm13111598
APA StyleMarciniak, A., Nawrocka-Rutkowska, J., Brodowska, A., Starczewski, A., & Szydłowska, I. (2023). Lymphangioleiomyomatosis with Tuberous Sclerosis Complex—A Case Study. Journal of Personalized Medicine, 13(11), 1598. https://doi.org/10.3390/jpm13111598