
Aicardi–Goutières Syndrome with Congenital Glaucoma Caused by Novel TREX1 Mutation


Abstract
:1. Introduction
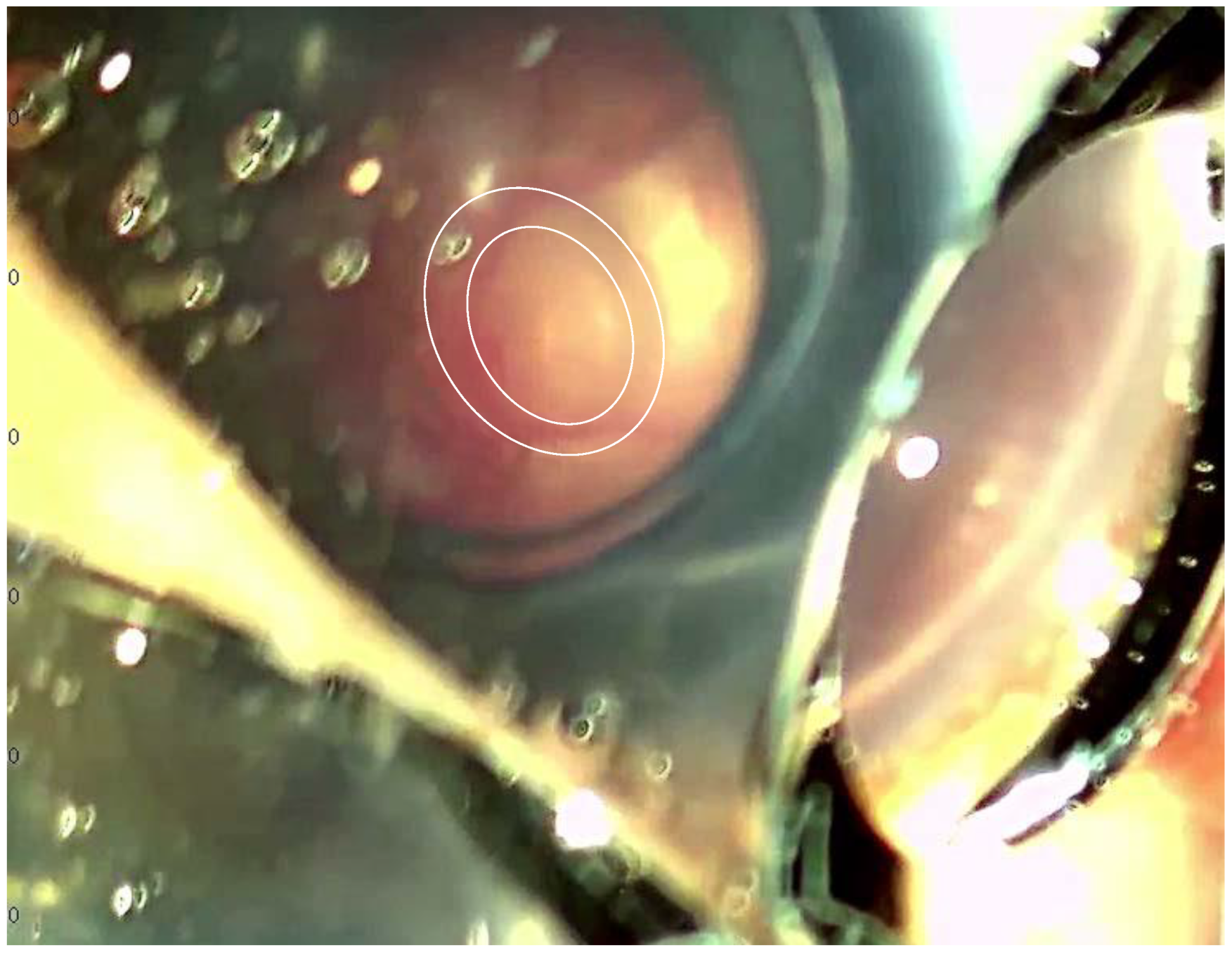
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crow, Y.J. Aicardi-Goutières syndrome. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 113, pp. 1629–1635. [Google Scholar] [CrossRef]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 2015, 2, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Adang, L.; Gavazzi, F.; De Simone, M.; Fazzi, E.; Galli, J.; Koh, J.; Kramer-Golinkoff, J.; De Giorgis, V.; Orcesi, S.; Peer, K.; et al. Developmental Outcomes of Aicardi Goutières Syndrome. J. Child Neurol. 2020, 35, 7–16. [Google Scholar] [CrossRef]
- Haaxma, C.A.; Crow, Y.J.; van Steensel, M.A.; Lammens, M.M.; Rice, G.I.; Verbeek, M.M.; Willemsen, M.A. A de novo p.Asp18Asn mutation in TREX1 in a patient with Aicardi-Goutières syndrome. Am. J. Med. Genet. A 2010, 10, 2612–2617. [Google Scholar] [CrossRef]
- Abe, J.; Nakamura, K.; Nishikomori, R.; Kato, M.; Mitsuiki, N.; Izawa, K.; Awaya, T.; Kawai, T.; Yasumi, T.; Toyoshima, I.; et al. A nationwide survey of Aicardi-Goutières syndrome patients identifies a strong association between dominant TREX1 mutations and chilblain lesions: Japanese cohort study. Rheumatology 2014, 3, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet. 2012, 11, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Orcesi, S.; La Piana, R.; Fazzi, E. Aicardi-Goutieres syndrome. Br. Med. Bull. 2009, 89, 183–201. [Google Scholar] [CrossRef]
- Livingston, J.H.; Stivaros, S.; van der Knaap, M.S.; Crow, Y.J. Recognizable phenotypes associated with intracranial calcification. Dev. Med. Child Neurol. 2013, 55, 46–57. [Google Scholar] [CrossRef]
- Maccora, I.; Marrani, E.; Mastrolia, M.V.; Abu-Rumeileh, S.; Maniscalco, V.; Fusco, E.; Barbati, F.; Pagnini, I.; Simonini, G. Ocular involvement in monogenic autoinflammatory disease. Autoimmun. Rev. 2021, 11, 102944. [Google Scholar] [CrossRef]
- Rice, G.I.; Forte, G.M.; Szynkiewicz, M.; Chase, D.S.; Aeby, A.; Abdel-Hamid, M.S.; Ackroyd, S.; Allcock, R.; Bailey, K.M.; Balottin, U.; et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A case-control study. Lancet Neurol. 2013, 12, 1159–1169. [Google Scholar] [CrossRef]
- Rice, G.; Patrick, T.; Parmar, R.; Taylor, C.F.; Aeby, A.; Aicardi, J.; Artuch, R.; Montalto, S.A.; Bacino, C.A.; Barroso, B.; et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 2007, 4, 713–725. [Google Scholar] [CrossRef]
- Crow, Y.J.; Livingston, J.H. Aicardi-Goutières syndrome: An important Mendelian mimic of congenital infection. Dev. Med. Child Neurol. 2008, 6, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Dell’Isola, G.B.; Dini, G.; Culpepper, K.L.; Portwood, K.E.; Ferrara, P.; Di Cara, G.; Verrotti, A.; Lodolo, M. Clinical spectrum and currently available treatment of type I interferonopathy Aicardi-Goutières syndrome. World J. Pediatr. 2023, 7, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Rodero, M.P.; Crow, Y.J. Human disease phenotypes associated with mutations in TREX1. J. Clin. Immunol. 2015, 3, 235–243. [Google Scholar] [CrossRef]
- Hemphill, W.O.; Perrino, F.W. Measuring TREX1 and TREX2 exonuclease activities. Methods Enzymol. 2019, 625, 109–133. [Google Scholar] [CrossRef]
- Yan, N. Immune Diseases Associated with TREX1 and STING Dysfunction. J. Interferon Cytokine Res. 2017, 5, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Jepps, H.; Seal, S.; Hattingh, L.; Crow, Y.J. The neonatal form of Aicardi-Goutières syndrome masquerading as congenital infection. Early Hum. Dev. 2008, 12, 783–785. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Massey, R.F.; Innes, J.R.; Pairaudeau, P.W.; Rowland Hill, C.A.; Woods, C.G.; Ali, M.; Livingstonz, J.H.; Lebon, P.; Nischall, K.; et al. Congenital glaucoma and brain stem atrophy as features of Aicardi-Goutières syndrome. Am. J. Med. Genet. A 2004, 3, 303–307. [Google Scholar] [CrossRef]
- Gowda, V.K.; Vegda, H.; Shivappa, S.K.; Benakappa, N. Aicardi-Goutieres Syndrome Presenting with Congenital Glaucoma. Indian J. Pediatr. 2020, 8, 652. [Google Scholar] [CrossRef]
- Musalem, H.M.; Dirar, Q.S.; Al-Hazzaa, S.A.F.; Al Zoba, A.A.; El-Mansoury, J. Unusual Association of Aniridia with Aicardi-Goutières Syndrome-Related Congenital Glaucoma in a Tertiary Care Center. Am. J. Case Rep. 2018, 19, 500–504. [Google Scholar] [CrossRef]
- Balikov, D.A.; Jacobson, A.; Prasov, L. Glaucoma Syndromes: Insights into Glaucoma Genetics and Pathogenesis from Monogenic Syndromic Disorders. Genes 2021, 9, 1403. [Google Scholar] [CrossRef]
- Lang, E.; Koller, S.; Atac, D.; Pfäffli, O.A.; Hanson, J.V.M.; Feil, S.; Bähr, L.; Bahr, A.; Kottke, R.; Joset, P.; et al. Genotype-phenotype spectrum in isolated and syndromic nanophthalmos. Acta Ophthalmol. 2021, 4, e594–e607. [Google Scholar] [CrossRef] [PubMed]
- Le Gargasson, J.F.; Rigaudiere, F.; Dufier, J.L.; Aicardi, J.; Goutieres, F.; Pizzato, M.; Grall, Y. Etude de l’électrophysiologie visuelle chez 101 enfants encéphalopathes. J. Fr. Ophtalmol. 1990, 8, 441–448. [Google Scholar]
- Ferreira, C.R.; Crow, Y.J.; Gahl, W.A.; Gardner, P.J.; Goldbach-Mansky, R.; Hur, S.; de Jesús, A.A.; Nehrebecky, M.; Park, J.W.; Briggs, T.A. DDX58 and Classic Singleton-Merten Syndrome. J. Clin. Immunol. 2019, 1, 75–80. [Google Scholar] [CrossRef] [PubMed]
- d’Angelo, D.M.; Di Filippo, P.; Breda, L.; Chiarelli, F. Type I Interferonopathies in Children: An Overview. Front. Pediatr. 2021, 9, 631329. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, F.; Singh, H.; Anand, N.; Ahmed, I.I. Intraocular pressure rise in the course of peginterferon alpha-2a, ribavirin, and boceprevir therapy for hepatitis C. Can. J. Ophthalmol. 2015, 6, e112-4. [Google Scholar] [CrossRef]
- Bagheri, H.; Fouladi, A.; Barange, K.; Lapeyre-Mestre, M.; Payen, J.L.; Montastruc, J.L.; Vinel, J.P. Follow-up of adverse drug reactions from peginterferon alfa-2b-ribavirin therapy. Pharmacotherapy 2004, 11, 1546–1553. [Google Scholar] [CrossRef]
- Kwon, Y.S.; Choe, Y.H.; Chin, H.S. Development of glaucoma in the course of interferon alpha therapy for chronic hepatitis B. Yonsei Med. J. 2001, 1, 134–136. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Hepatitis C virus: Standard-of-care treatment. Adv. Pharmacol. 2013, 67, 169–215. [Google Scholar] [CrossRef]
- Takase, H.; Sugita, S.; Rhee, D.J.; Imai, Y.; Taguchi, C.; Sugamoto, Y.; Tagawa, Y.; Nishihira, J.; Russell, P.; Mochizuki, M. The presence of macrophage migration inhibitory factor in human trabecular meshwork and its upregulatory effects on the T helper 1 cytokine. Investig. Ophthalmol. Vis. Sci. 2002, 8, 2691–2696. [Google Scholar]
- Chua, J.; Vania, M.; Cheung, C.M.; Ang, M.; Chee, S.P.; Yang, H.; Li, J.; Wong, T.T. Expression profile of inflammatory cytokines in aqueous from glaucomatous eyes. Mol. Vis. 2012, 18, 431–438. [Google Scholar]
- Kaneko, M.; Onishi, Y.; Okada, E.; Sasaki, Y.; Takahashi, K.; Aiba, S. Ultrasonographic diagnosis of fetal eye. Jpn. J. Clin. Ophthalmol. 1992, 46, 279–282. [Google Scholar]
- Kawabata, I. Evaluation of fetal anomaly using MRI. J. Jpn. Obstet. Gynecol. Soc. 2006, 58, 193–196. [Google Scholar]
- Tsukitome, H.; Uji, Y.; Yagi, T.; Sasoh, M. A case of developmental glaucoma suspected from megalophthalmos represented by obstetric ultrasonography. Jpn. J. Ophthalmol. 2011, 6, 686–687. [Google Scholar] [CrossRef] [PubMed]
- Rezaei Kanavi, M.; Yazdani, S.; Elahi, E.; Mirrahimi, M.; Hajizadeh, M.; Khodaverdi, S.; Suri, F. Prenatal diagnosis of primary congenital glaucoma and histopathological features in a fetal globe with cytochrome p4501B1 mutations. Eur. J. Ophthalmol. 2021, 3, 11206721211051235. [Google Scholar]
- Yu, Z.X.; Song, H.M. Toward a better understanding of type I interferonopathies: A brief summary, update and beyond. World J. Pediatr. 2020, 1, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Bienias, M.; Brück, N.; Griep, C.; Wolf, C.; Kretschmer, S.; Kind, B.; Tüngler, V.; Berner, R.; Lee-Kirsch, M.A. Therapeutic Approaches to Type I Interferonopathies. Curr. Rheumatol. Rep. 2018, 6, 32. [Google Scholar] [CrossRef]
- Kothur, K.; Bandodkar, S.; Chu, S.; Wienholt, L.; Johnson, A.; Barclay, P.; Brogan, P.A.; Rice, G.I.; Crow, Y.J.; Dale, R.C. An open-label trial of JAK 1/2 blockade in progressive IFIH1-associated neuroinflammation. Neurology 2018, 6, 289–291. [Google Scholar] [CrossRef]
- Rice, G.I.; Meyzer, C.; Bouazza, N.; Hully, M.; Boddaert, N.; Semeraro, M.; Zeef, L.A.H.; Rozenberg, F.; Bondet, V.; Duffy, D.; et al. Reverse-Transcriptase Inhibitors in the Aicardi–Goutières Syndrome. N. Engl. J. Med. 2018, 23, 2275–2277. [Google Scholar] [CrossRef]
- Wang, M.; Sooreshjani, M.A.; Mikek, C.; Opoku-Temeng, C.; Sintim, H.O. Suramin potently inhibits cGAMP synthase, cGAS, in THP1 cells to modulate IFN-β levels. Future Med. Chem. 2018, 11, 1301–1317. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Missense/Nonsense Mutation | |||
| HGMD Codon Change | HMD Amino Acid Change | HGVS (Nucleotide) | HGVS (Protein) |
| ACC-AAC | Thr13Asn | c.38C>A | p.T13N |
| ACC-CCC | Thr13Pro | c.37A>C | p.T13P |
| ATC-ATG | Ile15Met | c.45C>G | p.I15M |
| GAC-CAC | Asp18His | c.52G>C | p.D18H |
| ACG-AGG | Thr32Arg | c.95C>G | p.T32R |
| TGT-TGA | Cys42Term | c.126T>A | p.C42 |
| CCG-CAG | Pro61Gln | c.182C>A | p.P61Q |
| AAG-AGG | Lys66Arg | c.197A>G | p.K66R |
| CTG-CCG | Leu69Pro | c.206T>C | p.L69P |
| CTG-CAG | Leu92Gln | c.275T>A | p.L92Q |
| CGT-CAT | Arg97His | c.290G>A | p.R97H |
| CGC-CAC | Arg114His | c.341G>A | p.R114H |
| GTG-GCG | Val122Ala | c.365T>C | p.V122A |
| CTG-CCG | Leu162Pro | c.485T>C | p.L162P |
| CGA-TGA | Arg164Term | c.490C>T | p.R164 |
| CAC-TAC | His195Tyr | c.583C>T | p.H195Y |
| GAG-AAG | Glu198Lys | c.592G>A | p.E198K |
| GAT-AAT | Asp200Asn | c.598G>A | p.D200N |
| GAT-CAT | Asp200His | c.598G>C | p.D200H |
| GTC-GAC | Val201Asp | c.602T>A | p.V201D |
| TGG-TAG | Trp210Term | c.629G>A | p.W210 |
| GCC-ACC | Ala223Thr | c.667G>A | p.A223T |
| ACA-CCA | Thr303Pro | c.907A>C | p.T303P |
| Small deletions | |||
| HGMD deletion | HGVS (nucleotide) | HGVS (protein) | |
| CCCCC^49ACCTCtcAGGGGCCACC | c.150_151delTC | p.(Gln51Glyfs*50) | |
| CCCACC^50TCTCagGGGCCACCTC | c.152_153delAG | p.(Gln51Argfs*50) | |
| CCTGC^78AGCCCtgcagccAGCGAGATCA | c.237_243delTGCAGCC | p.(Ala81Argfs*5) | |
| GCCCT^80GCAGCcagcGAGATCACAG | c.243_246delCAGC | p.(Ser82Argfs*5) | |
| TACGAC^131TTCCcCCTGCTCCAA | c.397delC | p.(Leu133Cysfs*27) | |
| GTGGAT^155AGCAtCACTGCGCTG | c.467delT | p.(Ile156Thrfs*4) | |
| GATAGC^156ATCAcTGCGCTGAAG | c.470delC | p.(Thr157Metfs*3) | |
| CGAGCA^166AGCAgCCCCTCAGAA | c.500delG | p.(Ser167Thrfs*13) | |
| AGGAAG^176AGCTaTAGCCTAGGC | c.530delA | p.(Tyr177Leufs*3) | |
| GCTCAGC^207ATCtgtcaGTGGAGACCA | c.622_626delTGTCA | p.(Cys208Valfs*31) | |
| AGCCA^244AGACCaTCTGCTGTCA | c.735delA | p.(Ser246Leufs*31) | |
| AAGGAC^279CCTGgAGCCCTATCC | c.839delG | p.(Gly280Glufs*18) | |
| CCAGG^285GAGGGgCTGCTGGCCC | c.858delG | p.(Leu287Cysfs*11) | |
| GCTGCTG^289GCCccactgggtctgctggccATCCTGACCT | c.868_885del18 | p.(Pro290_Ala295del) | |
| Small insertions | |||
| HGMD insertion | HGVS (nucleotide) | HGVS (protein) | |
| TTTCGAC^19ATGgGAGGCCACTG | c.58dupG | p.(Glu20Glyfs*82) | |
| GAGCCCC^48CCCcACCTCTCAGG | c.144dupC | p.(Thr49Hisfs*53) | |
| CCTGTGT^71GTGtgGCTCCGGGGA | c.212_213dupTG | p.(Ala72Trpfs*17) | |
| CAGCCCT^80GCAaGCCAGCGAGA | c.240dupA | p.(Ala81Serfs*21) | |
| CCCTGCA^81GCCctgcagccAGCGAGATCA | c.236_243dupCTGCAGCC | p.(Ser82Leufs*9) | |
| ACAGGT^87CTGAagGCACAGCTGT | c.262_263insAG | p.(Ser88Lysfs*23) | |
| TGGGCGT^98CAAaTGTTTTGATG | c.294dupA | p.(Cys99Metfs*3) | |
| GTCAA^99TGTTTgtttTGATGACAAC | c.296_299dupGTTT | p.(Phe100Leufs*3) | |
| GCCTG^122GTGGCggcACACAATGGT | c.366_368dupGGC | p.(Ala123dup) | |
| GCTCCAA^136GCAccccctgctccaagcaGAGCTGGCTA | c.393_408dup16 | p.(Glu137Profs*24) | |
| TGAGGGT^200GATgatGTCCTGGCCC | c.599_601dupATG | p.(Asp200dup) | |
| ATCTGT^209CAGTcagtGGAGACCACA | c.625_628dupCAGT | p.(Trp210Serfs*32) | |
| TGTCAG^210TGGAtggaGACCACAGGC | c.628_631dupTGGA | p.(Arg211Metfs*31) | |
| CATCAGG^231CCCcATGTATGGGG | c.693dupC | p.(Met232Hisfs*9) | |
| Gross insertions | |||
| DNA level | Insertion/duplication | Description | |
| gDNA | duplication | 54 bp c.609_662 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Świerczyńska, M.; Tronina, A.; Filipek, E. Aicardi–Goutières Syndrome with Congenital Glaucoma Caused by Novel TREX1 Mutation. J. Pers. Med. 2023, 13, 1609. https://doi.org/10.3390/jpm13111609
Świerczyńska M, Tronina A, Filipek E. Aicardi–Goutières Syndrome with Congenital Glaucoma Caused by Novel TREX1 Mutation. Journal of Personalized Medicine. 2023; 13(11):1609. https://doi.org/10.3390/jpm13111609
Chicago/Turabian StyleŚwierczyńska, Marta, Agnieszka Tronina, and Erita Filipek. 2023. "Aicardi–Goutières Syndrome with Congenital Glaucoma Caused by Novel TREX1 Mutation" Journal of Personalized Medicine 13, no. 11: 1609. https://doi.org/10.3390/jpm13111609
APA StyleŚwierczyńska, M., Tronina, A., & Filipek, E. (2023). Aicardi–Goutières Syndrome with Congenital Glaucoma Caused by Novel TREX1 Mutation. Journal of Personalized Medicine, 13(11), 1609. https://doi.org/10.3390/jpm13111609