
Metagenomics Analysis of Breast Microbiome Highlights the Abundance of Rothia Genus in Tumor Tissues
 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling, Patients, and Ethics
2.2. Genomic DNA Extraction from Breast Tissue
2.3. Preparation of Samples and Sequencing of 16S-rRNA Amplicons
2.4. Sequencing Data Analysis
2.5. Statistical Analyses
2.6. Enterotype Clustering
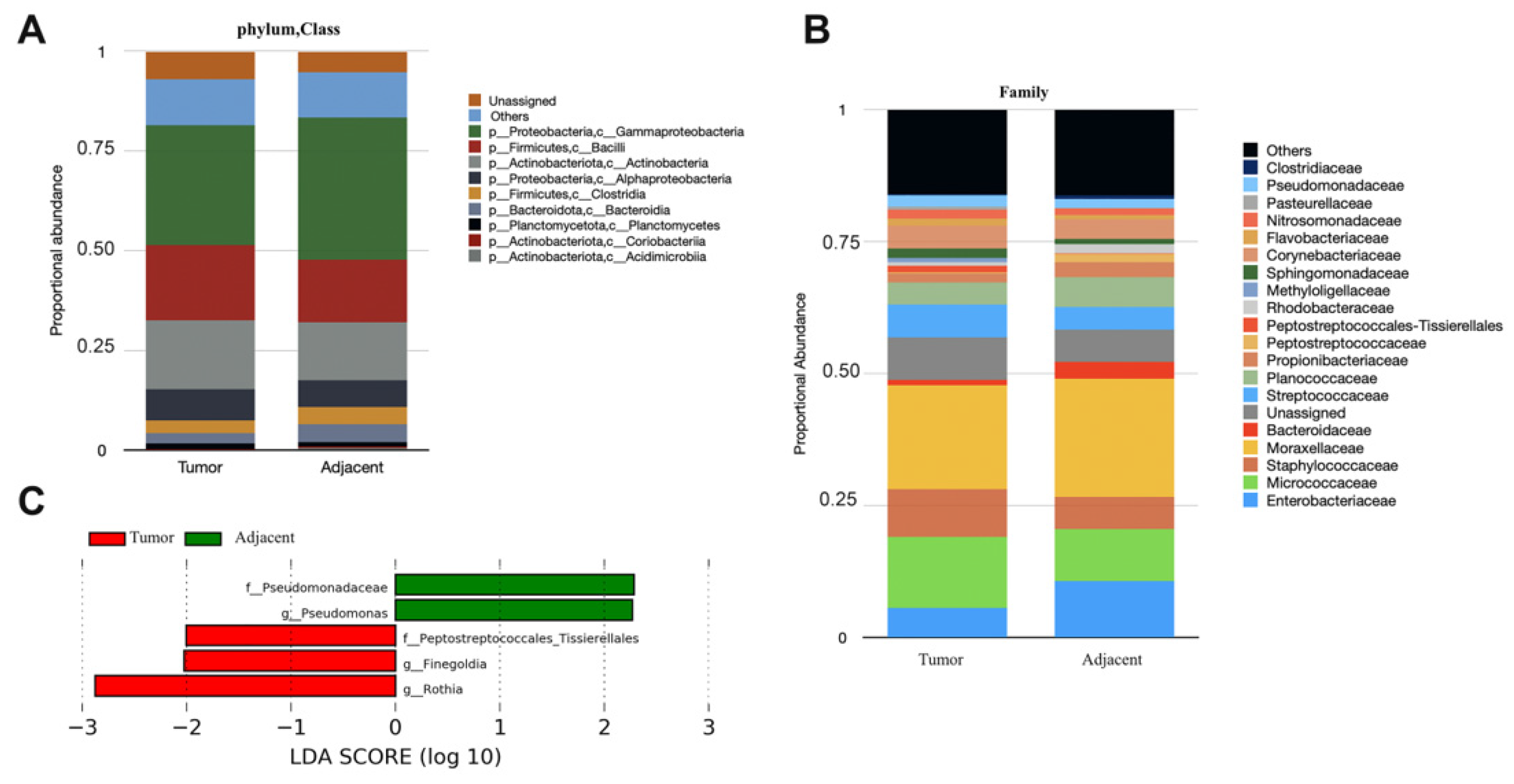
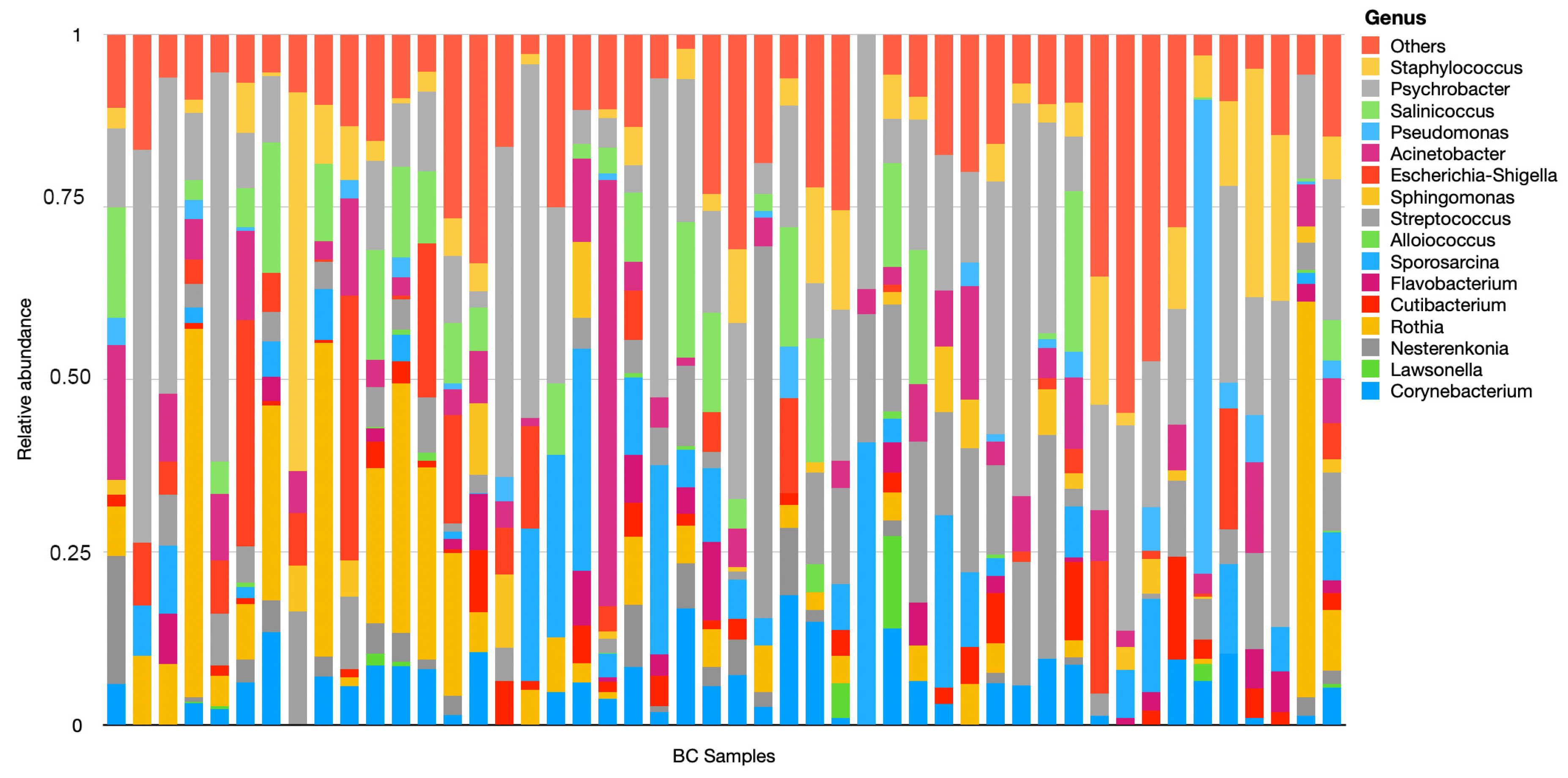
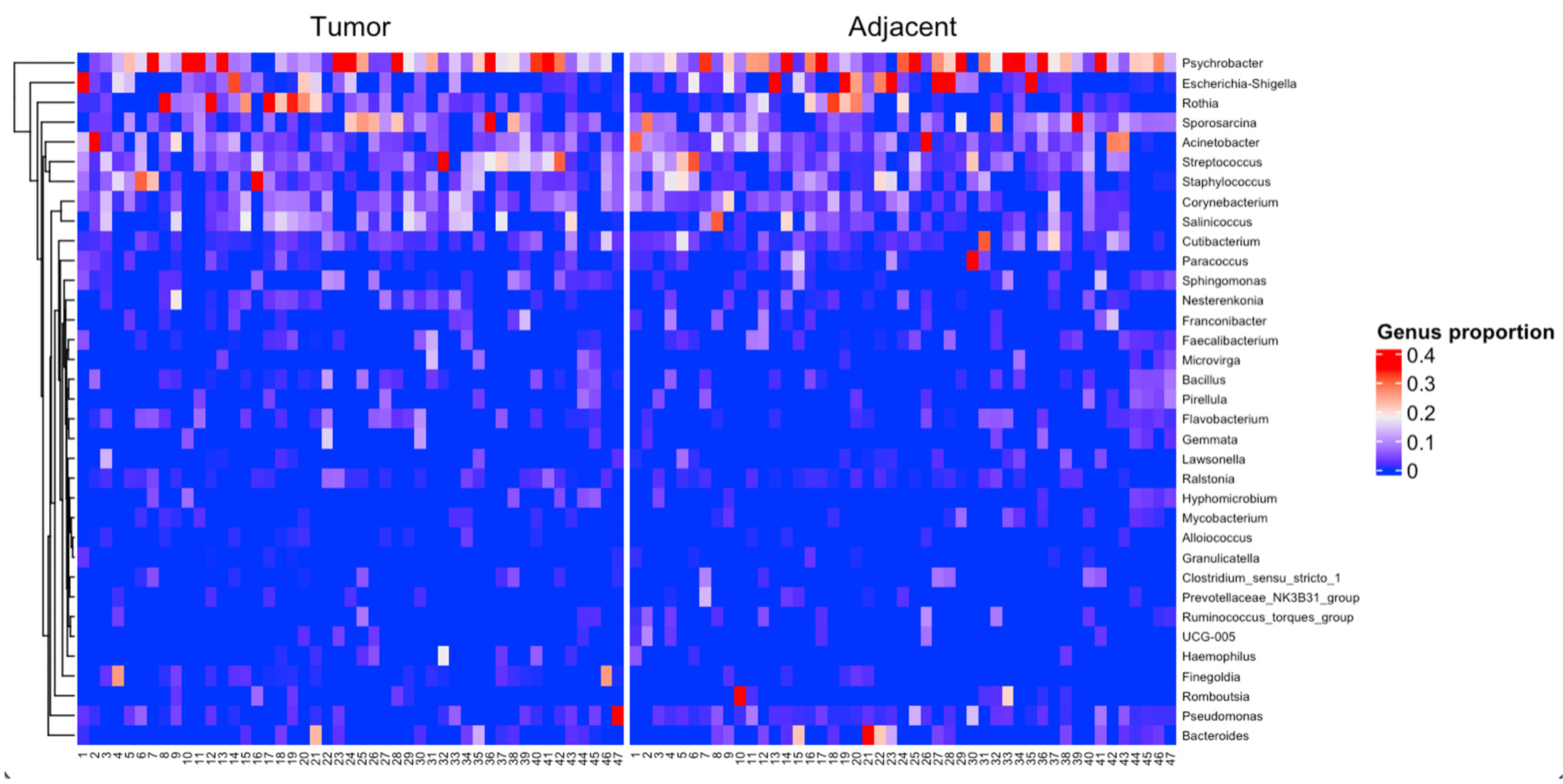
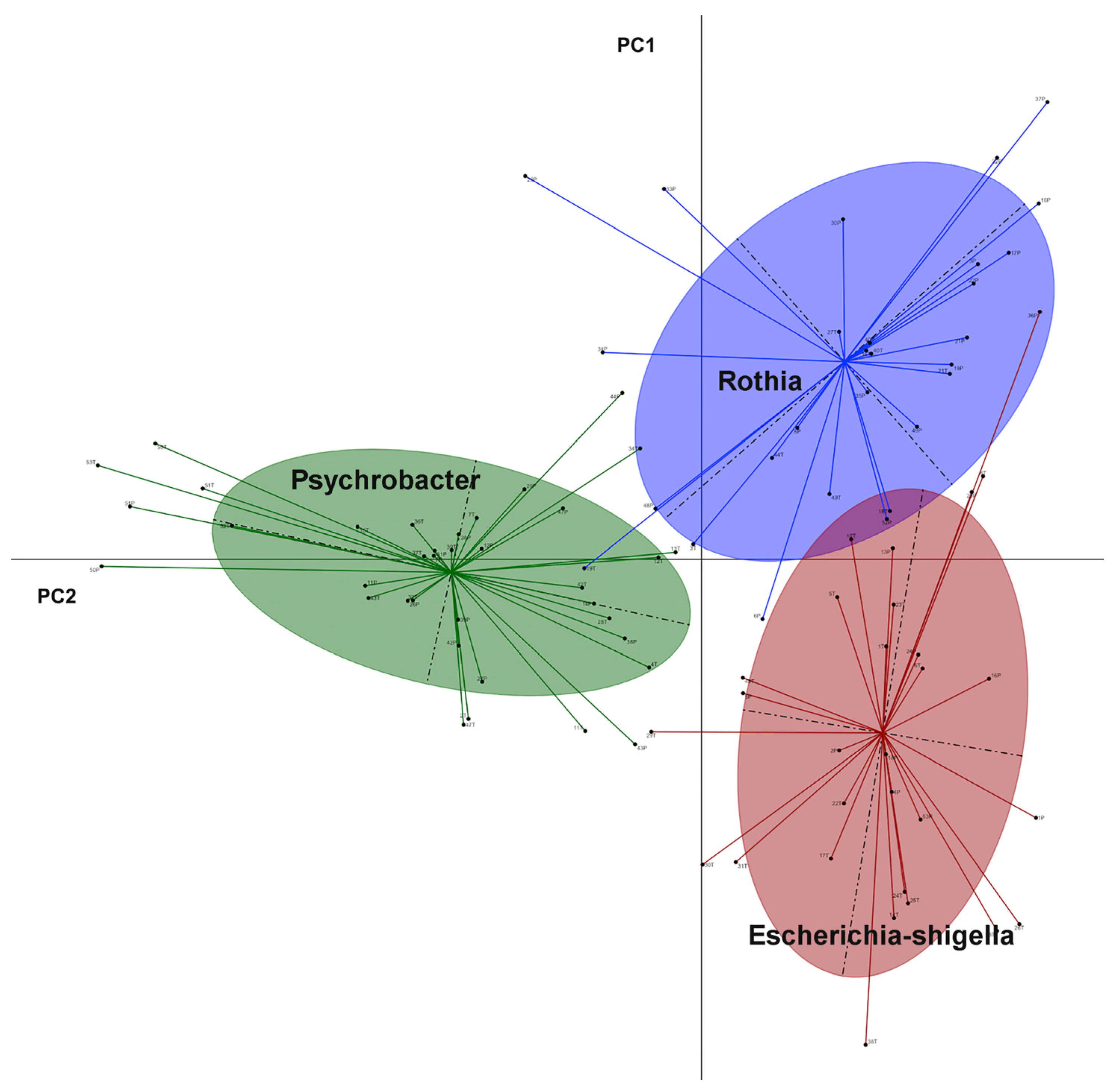
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chhikara, B.S.; Parang, K. Global Cancer Statistics 2022: The Trends Projection Analysis. Chem. Biol. Lett. 2022, 10, 1. [Google Scholar]
- Fernández, M.F.; Reina-Pérez, I.; Astorga, J.M.; Rodríguez-Carrillo, A.; Plaza-Díaz, J.; Fontana, L. Breast cancer and its relationship with the microbiota. Int. J. Environ. Res. Public Health 2018, 15, 1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costantini, L.; Magno, S.; Albanese, D.; Donati, C.; Molinari, R.; Filippone, A.; Masetti, R.; Merendino, N. Characterization of human breast tissue microbiota from core needle biopsies through the analysis of multi hypervariable 16S-rRNA gene regions. Sci. Rep. 2018, 8, 16893. [Google Scholar] [CrossRef] [Green Version]
- Felgner, S.; Kocijancic, D.; Frahm, M.; Weiss, S. Bacteria in Cancer Therapy: Renaissance of an Old Concept. Int. J. Microbiol. 2016, 2016, 8451728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekaboruah, E.; Suryavanshi, M.V.; Chettri, D.; Verma, A.K. Human microbiome: An academic update on human body site specific surveillance and its possible role. Arch. Microbiol. 2020, 202, 2147–2167. [Google Scholar] [CrossRef] [PubMed]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef]
- Chadha, J.; Nandi, D.; Atri, Y.; Nag, A. Significance of human microbiome in breast cancer: Tale of an invisible and an invincible. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Karpiński, T.M.; Adamczak, A. Anticancer Activity of Bacterial Proteins and Peptides. Pharmaceutics 2018, 10, 54. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.N.; Falk, R.T.; Schairer, C.; Moore, S.C.; Fuhrman, B.J.; Dallal, C.M.; Bauer, D.C.; Dorgan, J.F.; Shu, X.-O.; Zheng, W. Association of Estrogen Metabolism with Breast Cancer Risk in Different Cohorts of Postmenopausal WomenEstrogen Metabolism and Breast Cancer Risk. Cancer Res. 2017, 77, 918–925. [Google Scholar] [CrossRef] [Green Version]
- Xuan, C.; Shamonki, J.M.; Chung, A.; DiNome, M.L.; Chung, M.; Sieling, P.A.; Lee, D.J. Microbial dysbiosis is associated with human breast cancer. PLoS ONE 2014, 9, e83744. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Altemus, J.; Niazi, F.; Green, H.; Calhoun, B.C.; Sturgis, C.; Grobmyer, S.R.; Eng, C. Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget 2017, 8, 88122. [Google Scholar] [CrossRef] [Green Version]
- Urbaniak, C.; Gloor, G.B.; Brackstone, M.; Scott, L.; Tangney, M.; Reid, G. The microbiota of breast tissue and its association with breast cancer. Appl. Environ. Microbiol. 2016, 82, 5039–5048. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef] [Green Version]
- Rea, D.; Coppola, G.; Palma, G.; Barbieri, A.; Luciano, A.; Del Prete, P.; Rossetti, S.; Berretta, M.; Facchini, G.; Perdonà, S. Microbiota effects on cancer: From risks to therapies. Oncotarget 2018, 9, 17915. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Urbaniak, C.; Cummins, J.; Brackstone, M.; Macklaim, J.M.; Gloor, G.B.; Baban, C.K.; Scott, L.; O'Hanlon, D.M.; Burton, J.P.; Francis, K.P. Microbiota of human breast tissue. Appl. Environ. Microbiol. 2014, 80, 3007–3014. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Sun, T.; Xu, J. Tumor-related microbiome in the breast microenvironment and breast cancer. J. Cancer 2021, 12, 4841. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.N.; Istiaq, A.; Rahman, M.S.; Islam, M.R.; Anwar, A.; Siddiki, A.M.A.M.Z.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Microbiome dynamics and genomic determinants of bovine mastitis. Genomics 2020, 112, 5188–5203. [Google Scholar] [CrossRef] [PubMed]
- Hieken, T.J.; Chen, J.; Hoskin, T.L.; Walther-Antonio, M.; Johnson, S.; Ramaker, S.; Xiao, J.; Radisky, D.C.; Knutson, K.L.; Kalari, K.R.; et al. The Microbiome of Aseptically Collected Human Breast Tissue in Benign and Malignant Disease. Sci. Rep. 2016, 6, 30751. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of high-fat diet on gut microbiota: A driving force for chronic disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Gail, M.H.; Consonni, D.; Carugno, M.; Humphrys, M.; Pesatori, A.C.; Caporaso, N.E.; Goedert, J.J.; Ravel, J.; Landi, M.T. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol. 2016, 17, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, A.; Garrity, G.M. Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 10. [Google Scholar] [CrossRef] [PubMed]
- Panis, C.; Victorino, V.J.; Herrera, A.C.; Freitas, L.F.; De Rossi, T.; Campos, F.C.; Simão, A.N.; Barbosa, D.S.; Pinge-Filho, P.; Cecchini, R.; et al. Differential oxidative status and immune characterization of the early and advanced stages of human breast cancer. Breast Cancer Res. Treat. 2012, 133, 881–888. [Google Scholar] [CrossRef]
- Leek, R.D.; Landers, R.; Fox, S.B.; Ng, F.; Harris, A.L.; Lewis, C.E. Association of tumour necrosis factor alpha and its receptors with thymidine phosphorylase expression in invasive breast carcinoma. Br. J. Cancer 1998, 77, 2246–2251. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Akhtar, M.; Chen, Y.; Ma, Z.; Liang, Y.; Shi, D.; Cheng, R.; Cui, L.; Hu, Y.; Nafady, A.A.; et al. Chicken jejunal microbiota improves growth performance by mitigating intestinal inflammation. Microbiome 2022, 10, 107. [Google Scholar] [CrossRef]
- Chan, A.; Bashir, M.; Rivas, M.; Duvall, K.; Sieling, P.; Pieber, T.; Vaishampayan, P.; Love, S.; Lee, D. Characterization of the microbiome of nipple aspirate fluid of breast cancer survivors. Sci. Rep. 2016, 6, 28061. [Google Scholar] [CrossRef] [Green Version]
- Parida, S.; Sharma, D. The power of small changes: Comprehensive analyses of microbial dysbiosis in breast cancer. Biochim. Et. Biophys. Acta (BBA)-Rev. Cancer 2019, 1871, 392–405. [Google Scholar] [CrossRef]
- Bisgaard, H.; Li, N.; Bonnelykke, K.; Chawes, B.L.; Skov, T.; Paludan-Müller, G.; Stokholm, J.; Smith, B.; Krogfelt, K.A. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J. Allergy Clin. Immunol. 2011, 128, 646–652.e1–e5. [Google Scholar] [CrossRef]
- Bull, M.; Plummer, S.; Marchesi, J.; Mahenthiralingam, E. The life history of Lactobacillus acidophilus as a probiotic: A tale of revisionary taxonomy, misidentification and commercial success. FEMS Microbiol. Lett. 2013, 349, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Zai, H.; Zhang, K.; Zhang, X.; Luo, N.; Li, X.; Hu, Y.; Wu, Y. L-norvaline affects the proliferation of breast cancer cells based on the microbiome and metabolome analysis. J. Appl. Microbiol. 2022, 133, 1014–1026. [Google Scholar] [CrossRef]
- Ming, Z.; Zou, Z.; Cai, K.; Xu, Y.i.; Chen, X.; Yi, W.; Luo, J.; Luo, Z. ARG1 functions as a tumor suppressor in breast cancer. Acta Biochim. Et Biophys. Sin. 2020, 52, 1257–1264. [Google Scholar] [CrossRef]
- Liu, J.; Williams, B.; Frank, D.; Dillon, S.M.; Wilson, C.C.; Landay, A.L. Inside out: HIV, the gut microbiome, and the mucosal immune system. J. Immunol. 2017, 198, 605–614. [Google Scholar]
- Muccee, F.; Ghazanfar, S.; Ajmal, W.; Al-Zahrani, M. In-Silico Characterization of Estrogen Reactivating & beta;-Glucuronidase Enzyme in GIT Associated Microbiota of Normal Human and Breast Cancer Patients. Genes 2022, 13, 1545. [Google Scholar]
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J. Biol. Chem. 2019, 294, 18586–18599. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 16s Region | Name | F/R | Sequence |
|---|---|---|---|
| V3-V4 [15] | S-D-Bact-0341-b-S-17 | F | 5′ TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG |
| S-D-Bact-0785-a-A-21 | R | 5′ GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA G GAC TAC HVG GGT ATC TAA TCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kartti, S.; Bendani, H.; Boumajdi, N.; Bouricha, E.M.; Zarrik, O.; EL Agouri, H.; Fokar, M.; Aghlallou, Y.; EL Jaoudi, R.; Belyamani, L.; et al. Metagenomics Analysis of Breast Microbiome Highlights the Abundance of Rothia Genus in Tumor Tissues. J. Pers. Med. 2023, 13, 450. https://doi.org/10.3390/jpm13030450
Kartti S, Bendani H, Boumajdi N, Bouricha EM, Zarrik O, EL Agouri H, Fokar M, Aghlallou Y, EL Jaoudi R, Belyamani L, et al. Metagenomics Analysis of Breast Microbiome Highlights the Abundance of Rothia Genus in Tumor Tissues. Journal of Personalized Medicine. 2023; 13(3):450. https://doi.org/10.3390/jpm13030450
Chicago/Turabian StyleKartti, Souad, Houda Bendani, Nasma Boumajdi, El Mehdi Bouricha, Oumaima Zarrik, Hajar EL Agouri, Mohamed Fokar, Youssef Aghlallou, Rachid EL Jaoudi, Lahcen Belyamani, and et al. 2023. "Metagenomics Analysis of Breast Microbiome Highlights the Abundance of Rothia Genus in Tumor Tissues" Journal of Personalized Medicine 13, no. 3: 450. https://doi.org/10.3390/jpm13030450