
A Comprehensive Analysis of Programmed Cell Death-Associated Genes for Tumor Microenvironment Evaluation Promotes Precise Immunotherapy in Patients with Lung Adenocarcinoma
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Study Design
2.2. Construction and Validation of the Programmed Cell Death Associated-Gene Models
2.3. Evaluation of the Tumor Microenvironment and Immunotherapy Response
2.4. Cell Culture
2.5. RNA Isolation, cDNA Synthesis, and qPCR Analysis
2.6. Statistical Analysis
3. Results
3.1. Identification of Apoptosis-, Ferroptosis-, and Autophagy-Associated Differentially Expressed Genes
3.2. Apoptosis-Associated Gene Risk Score Associated with Survival
3.3. Ferroptosis-Associated Gene Risk Score Associated with Survival
3.4. Autophagy-Associated Gene Risk Score Associated with Survival
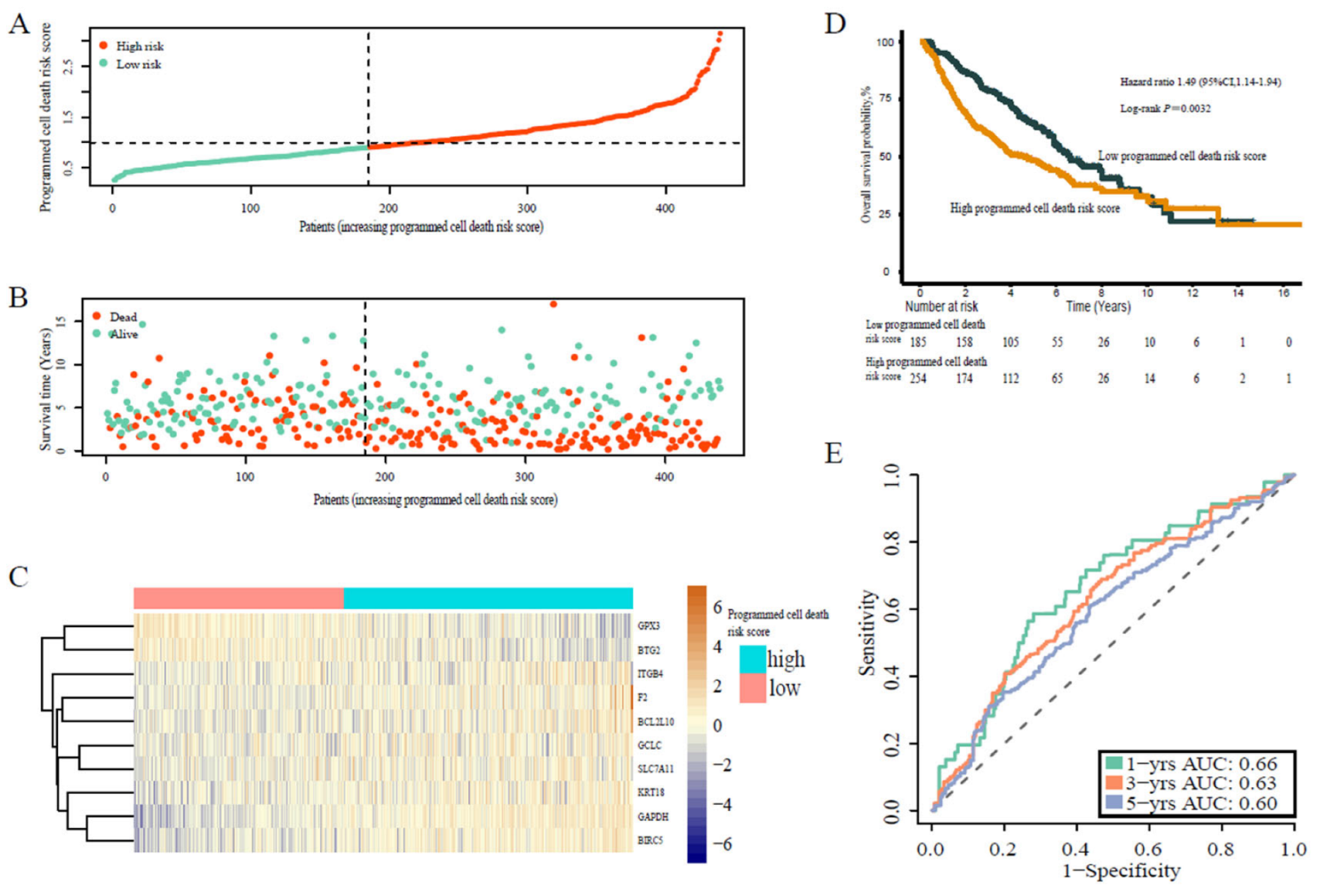
3.5. Programmed Cell Death-Associated Gene Risk Score Associated with Survival
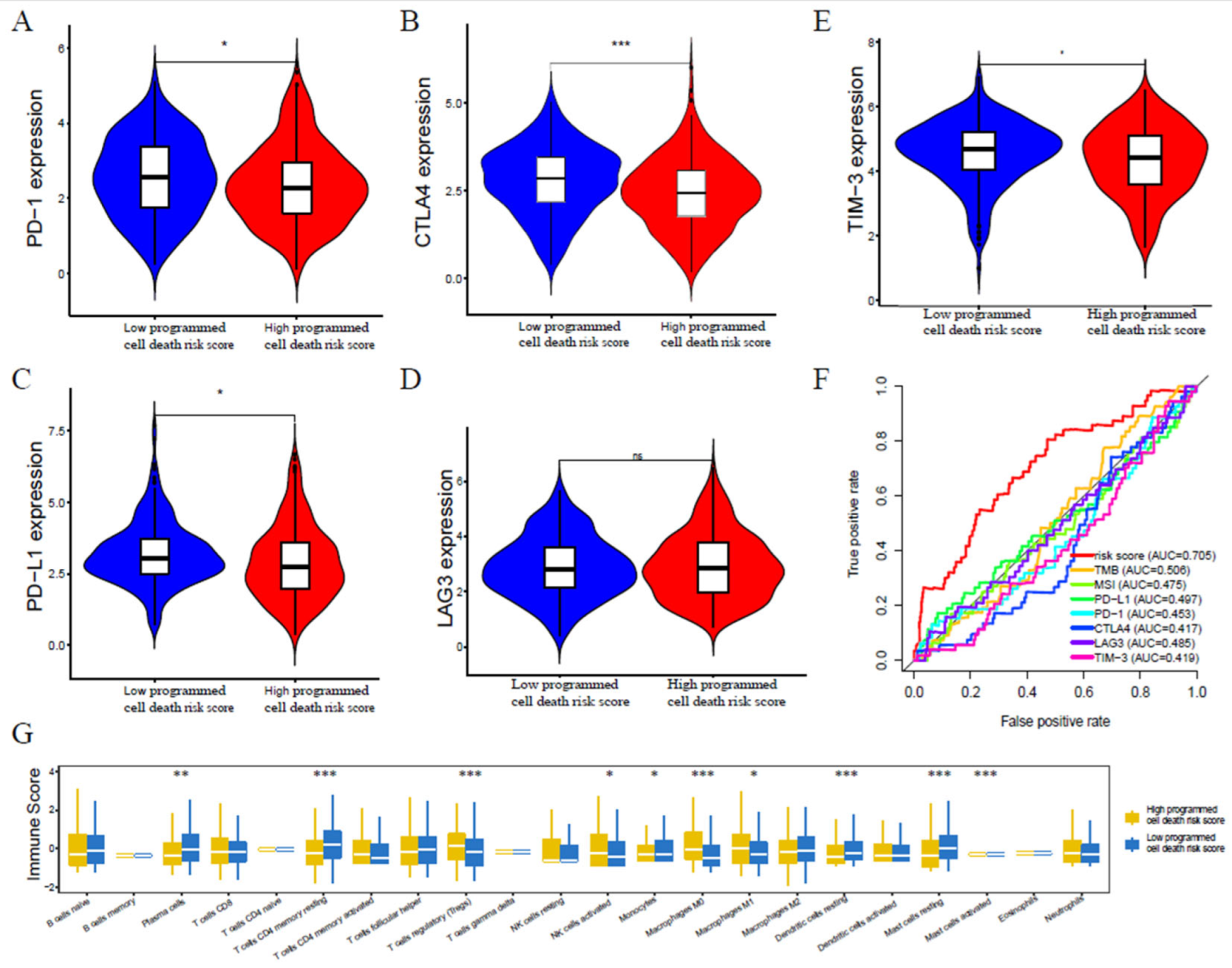
3.6. Programmed Cell Death-Associated Gene Risk Score Associated with Tumor Immune Microenvironment
3.7. Programmed Cell Death-Associated Gene Risk Score Associated with Immunotherapy Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Chen, Y.; Zhang, W.; Zhong, H.; Ou, Q.; Gu, Y.; Xia, J.; Lin, D.; Fu, T.; Liu, R.; et al. Joint association of patients’ sex and PD-L1 expression with overall survival benefits and tumor-immune microenvironment in immune checkpoint inhibitors for cancers. Clin. Transl. Med. 2020, 10, e92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meléndez, B.; Van Campenhout, C.; Rorive, S.; Remmelink, M.; Salmon, I.; D’Haene, N. Methods of measurement for tumor mutational burden in tumor tissue. Transl. Lung Cancer Res. 2018, 7, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Elkord, E. Treg-mediated acquired resistance to immune checkpoint inhibitors. Cancer Lett. 2019, 457, 168–179. [Google Scholar] [CrossRef]
- Wood, M.A.; Weeder, B.; David, J.; Nellore, A.; Thompson, R.F. Burden of tumor mutations, neoepitopes, and other variants are weak predictors of cancer immunotherapy response and overall survival. Genome Med. 2020, 12, 33. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Zeng, D.; Ou, Q.; Liu, S.; Li, A.; Chen, Y.; Lin, D.; Gao, Q.; Zhou, H.; Liao, W.; et al. Association of Survival and Immune-Associated Biomarkers With Immunotherapy in Patients With Non-Small Cell Lung Cancer: A Meta-analysis and Individual Patient-Level Analysis. JAMA Netw. Open 2019, 2, e196879. [Google Scholar] [CrossRef] [Green Version]
- Dercle, L.; Fronheiser, M.; Lu, L.; Du, S.; Hayes, W.; Leung, D.K.; Roy, A.; Wilkerson, J.; Guo, P.; Fojo, A.T.; et al. Identification of Non–Small Cell Lung Cancer Sensitive to Systemic Cancer Therapies Using Radiomics. Clin. Cancer Res. 2020, 26, 2151–2162. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Chen, Y.; Li, A.; Ou, Q.; Li, Q.; Gu, Y.; Lin, D.; Zhang, W.; Wang, J.; Tang, X.; et al. Novel blood-based tumor mutation algorithm and nomogram predict survival of immune checkpoint inhibitor in non-small-cell lung cancer: Results from two multicenter, randomized clinical trials. Clin. Transl. Med. 2020, 10, e53. [Google Scholar] [CrossRef]
- Hotchkiss, R.; Strasser, A.; McDunn, J.; Swanson, P.E. Cell death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.; Shah, R.; Aldrovandi, M.; da Silva, M.; Ingold, I.; Grocin, A.G.; da Silva, T.X.; Panzilius, E.; Scheel, C.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar]
- Hassannia, B.; Vandenabeele, P.; Berghe, T.V. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Stockwell, B.; Angeli, J.F.; Bayir, H.; Bush, A.; Conrad, M.; Dixon, S.; Fulda, S.; Gascón, S.; Hatzios, S.; Kagan, V.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Malta, T.; Sokolov, A.; Gentles, A.; Burzykowski, T.; Poisson, L.; Weinstein, J.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e15. [Google Scholar] [CrossRef] [Green Version]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Xu, W.; Zhang, L.; Liu, T.; Jin, G.; Song, J.; Wu, J.; Wang, Y.; Chen, W.; Zhang, C.; et al. Development and validation of a CIMP-associated prognostic model for hepatocellular carcinoma. Ebiomedicine 2019, 47, 128–141. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef]
- Fridman, W.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Zeng, D.; Li, M.; Zhou, R.; Zhang, J.; Sun, H.; Shi, M.; Bin, J.; Liao, Y.; Rao, J.; Liao, W. Tumor Microenvironment Characterization in Gastric Cancer Identifies Prognostic and Immunotherapeutically Relevant Gene Signatures. Cancer Immunol. Res. 2019, 7, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Zeng, D.; Ye, Z.; Wu, J.; Zhou, R.; Fan, X.; Wang, G.; Huang, Y.; Wu, J.; Sun, H.; Wang, M.; et al. Macrophage correlates with immunophenotype and predicts anti-PD-L1 response of urothelial cancer. Theranostics 2020, 10, 7002–7014. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lin, D.; Li, A.; Chen, Y.; Ou, Q.; Hu, H.; Yao, H. Association of Immune Checkpoint Inhibitor Therapy With Survival in Patients With Cancers With MUC16 Variants. JAMA Netw. Open 2020, 3, e205837. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Li, A.; Lin, D.; Gu, Y.; Chen, Y.; Ou, Q.; Li, L.; Yao, H.; Yu, Y. Association of immune checkpoint inhibitor with survival in patients with cancers with protein tyrosine phosphatase receptor T mutation. Clin. Transl. Med. 2020, 10, e214. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, W.; Anlin, L.; Chen, Y.; Ou, Q.; He, Z.; Zhang, Y.; Liu, R.; Yao, H.; Song, E. Association of Long Noncoding RNA Biomarkers With Clinical Immune Subtype and Prediction of Immunotherapy Response in Patients With Cancer. JAMA Netw. Open 2020, 3, e202149. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Lin, L.; Qin, T.; Ren, W.; Tan, Y.; Yang, Q.; Xu, H.; Xie, X.; Chen, Y.; Liu, S.; et al. Neoadjuvant camrelizumab, nab-paclitaxel, and carboplatin in patients with stage IB-IIIA non-small cell lung cancer (NANE-LC): A study protocol of prospective, single-arm, multicenter, phase II study. J. Thorac. Dis. 2021, 13, 6468–6475. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Ouyang, W.; Wang, Z.; Huang, H.; Ou, Q.; Lin, R.; Yu, Y.; Yao, H. A Comprehensive Analysis of Programmed Cell Death-Associated Genes for Tumor Microenvironment Evaluation Promotes Precise Immunotherapy in Patients with Lung Adenocarcinoma. J. Pers. Med. 2023, 13, 476. https://doi.org/10.3390/jpm13030476
Huang Y, Ouyang W, Wang Z, Huang H, Ou Q, Lin R, Yu Y, Yao H. A Comprehensive Analysis of Programmed Cell Death-Associated Genes for Tumor Microenvironment Evaluation Promotes Precise Immunotherapy in Patients with Lung Adenocarcinoma. Journal of Personalized Medicine. 2023; 13(3):476. https://doi.org/10.3390/jpm13030476
Chicago/Turabian StyleHuang, Yunxi, Wenhao Ouyang, Zehua Wang, Hong Huang, Qiyun Ou, Ruichong Lin, Yunfang Yu, and Herui Yao. 2023. "A Comprehensive Analysis of Programmed Cell Death-Associated Genes for Tumor Microenvironment Evaluation Promotes Precise Immunotherapy in Patients with Lung Adenocarcinoma" Journal of Personalized Medicine 13, no. 3: 476. https://doi.org/10.3390/jpm13030476