
Prevention of Protease-Induced Degradation of Desmoplakin via Small Molecule Binding

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Purification
2.2. Drug Library
2.3. Fluorescence Polarization Assays
2.4. Calpain Assays
2.5. Molecular Dynamics
2.6. Statistics
3. Results
3.1. Fluorescence Polarization Assays Can Monitor DSP Degradation
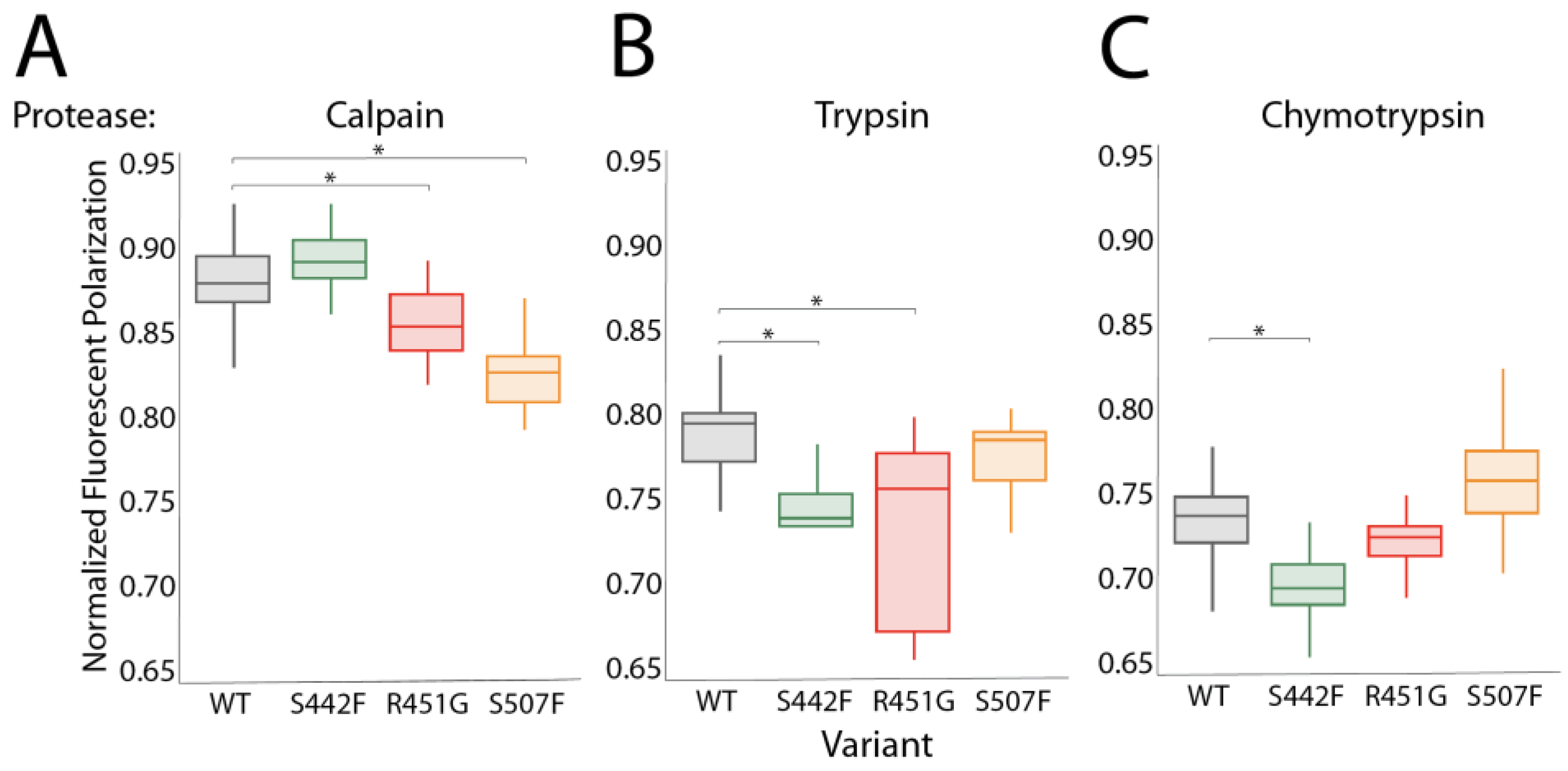
3.2. Proteases Other Than Calpain Can Hyperdegrade DSP Variants
3.3. Many Drugs Can Prevent DSP-Specific Degradation In Vitro
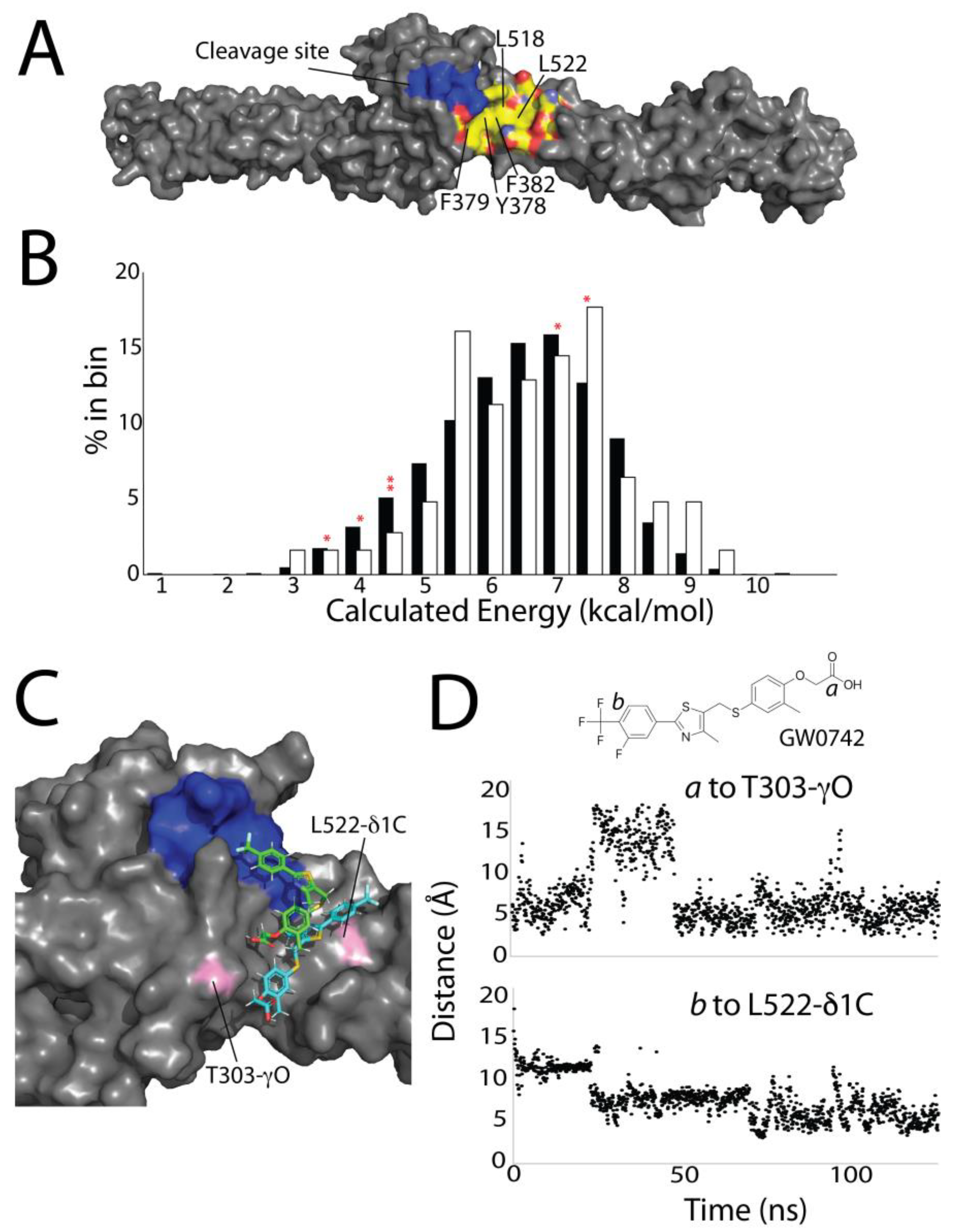
3.4. Computational Screening Identifies a Shallow Hydrophobic Cleft Where Drugs May Bind
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 785–802. [Google Scholar] [CrossRef]
- Corrado, D.; Wichter, T.; Link, M.S.; Hauer, R.N.W.; Marchlinski, F.E.; Anastasakis, A.; Bauce, B.; Basso, C.; Brunckhorst, C.; Tsatsopoulou, A.; et al. Treatment of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: An International Task Force Consensus Statement. Circulation 2015, 132, 441–453. [Google Scholar] [CrossRef]
- Akdis, D. Arrhythmogenic Cardiomyopathy: Electrical and Structural Phenotypes. Arrhythm. Electrophysiol. Rev. 2016, 5, 90–101. [Google Scholar] [CrossRef]
- Chua, C.J.; Morrissette-McAlmon, J.; Tung, L.; Boheler, K.R. Understanding Arrhythmogenic Cardiomyopathy: Advances through the use of Human Pluripotent Stem Cell Models. Genes 2023, 14, 1864. [Google Scholar] [CrossRef]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular Mechanisms of Arrhythmogenic Cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Migliore, F.; Mattesi, G.; Zorzi, A.; Bauce, B.; Rigato, I.; Corrado, D.; Cipriani, A. Arrhythmogenic Cardiomyopathy—Current Treatment and Future Options. J. Clin. Med. 2021, 10, 2750. [Google Scholar] [CrossRef]
- Calkins, H.; Corrado, D.; Marcus, F. Risk Stratification in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2017, 136, 2068–2082. [Google Scholar] [CrossRef]
- DeWitt, E.S.; Chandler, S.F.; Hylind, R.J.; Beausejour Ladouceur, V.; Blume, E.D.; VanderPluym, C.; Powell, A.J.; Fynn-Thompson, F.; Roberts, A.E.; Sanders, S.P.; et al. Phenotypic Manifestations of Arrhythmogenic Cardiomyopathy in Children and Adolescents. J. Am. Coll. Cardiol. 2019, 74, 346–358. [Google Scholar] [CrossRef]
- Hylind, R.; Beauséjour-Ladouceur, V.; Plovanich, M.E.; Helms, A.; Smith, E.; Joyce, E.; Granter, S.; Stevenson, L.W.; Cirino, A.L.; McDonough, B.A.; et al. Cardiocutaneous Features of Autosomal Dominant Desmoplakin-Associated Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, E003081. [Google Scholar] [CrossRef]
- Mohammed, F.; Chidgey, M. Desmosomal Protein Structure and Function and the Impact of Disease-causing Mutations. J. Struct. Biol. 2021, 213, 107749. [Google Scholar] [CrossRef]
- Hoorntje, E.T.; Te Rijdt, W.P.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; Van Tintelen, J.P. Arrhythmogenic Cardiomyopathy: Pathology, Genetics, and Concepts in Pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef]
- Yoder, M.W.; Wright, N.T.; Borzok, M.A. Calpain Regulation and Dysregulation—Its Effects on the Intercalated Disk. Int. J. Mol. Sci. 2023, 24, 11726. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Czarnowska, E.; Barbera, M.D.; Bauce, B.; Beffagna, G.; Wlodarska, E.K.; Pilichou, K.; Ramondo, A.; Lorenzon, A.; Wozniek, O.; et al. Ultrastructural Evidence of Intercalated Disc Remodelling in Arrhythmogenic Right Ventricular Cardiomyopathy: An Electron Microscopy Investigation on Endomyocardial Biopsies. Eur. Heart J. 2006, 27, 1847–1854. [Google Scholar] [CrossRef]
- Su, W.; van Wijk, S.W.; Brundel, B.J.J.M. Desmin variants: Trigger for Cardiac Arrhythmias? Front. Cell. Dev. Biol. 2022, 9, 986718. [Google Scholar] [CrossRef]
- Estigoy, C.B.; Pontén, F.; Odeberg, J.; Herbert, B.; Guilhaus, M.; Charleston, M.; Ho, J.W.K.; Cameron, D.; dos Remedios, C.G. Intercalated Discs: Multiple Proteins Perform Multiple Functions in Non-Failing and Failing Human Hearts. Biophys. Rev. 2009, 1, 43–49. [Google Scholar] [CrossRef]
- Sato, P.Y.; Coombs, W.; Lin, X.; Nekrasova, O.; Green, K.J.; Isom, L.L.; Taffet, S.M.; Delmar, M. Interactions between Ankyrin-G, Plakophilin-2, and Connexin43 at the Cardiac Intercalated Disc. Circ. Res. 2011, 109, 193–201. [Google Scholar] [CrossRef]
- Ng, R.; Manring, H.; Papoutsidakis, N.; Albertelli, T.; Tsai, N.; See, C.J.; Li, X.; Park, J.; Stevens, T.L.; Bobbili, P.J.; et al. Patient Mutations Linked to Arrhythmogenic Cardiomyopathy Enhance Calpain-Mediated Desmoplakin Degradation. JCI Insight 2019, 4, 128643. [Google Scholar] [CrossRef]
- Uzumcu, A.; Norgett, E.E.; Dindar, A.; Uyguner, O.; Nisli, K.; Kayserili, H.; Sahin, S.E.; Dupont, E.; Severs, N.J.; Leigh, I.M.; et al. Loss of Desmoplakin Isoform I Causes Early Onset Cardiomyopathy and Heart Failure in a Naxos-like Syndrome. J. Med. Genet. 2006, 43, e05. [Google Scholar] [CrossRef] [PubMed]
- Alcalai, R.; Metzger, S.; Rosenheck, S.; Meiner, V.; Chajek-Shaul, T. A Recessive Mutation in Desmoplakin Causes Arrhythmogenic Right Ventricular Dysplasia, Skin Disorder, and Woolly Hair. J. Am. Coll. Cardiol. 2003, 42, 319–327. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in Human Desmoplakin Domain Binding to Plakoglobin Causes a Dominant Form of Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef]
- Stevens, T.L.; Wallace, M.J.; El Refaey, M.; Roberts, J.D.; Koenig, S.N.; Mohler, P.J. Arrhythmogenic Cardiomyopathy: Molecular Insights for Improved Therapeutic Design. J. Cardiovasc. Dev. Dis. 2020, 7, 21. [Google Scholar] [CrossRef]
- Kang, H.; Weiss, T.M.; Bang, I.; Weis, W.I.; Choi, H.J. Structure of the Intermediate Filament-Binding Region of Desmoplakin. PLoS ONE 2016, 11, e0147641. [Google Scholar] [CrossRef]
- Choi, H.J.; Park-Snyder, S.; Pascoe, L.T.; Green, K.J.; Weis, W.I. Structures of Two Intermediate Filament-Binding Fragments of Desmoplakin Reveal a Unique Repeat Motif Structure. Nat. Struct. Biol. 2002, 9, 612–620. [Google Scholar] [CrossRef]
- Mueller, L.; Hatzfeld, M.; Keil, R. Desmosomes as Signaling Hubs in the Regulation of Cell Behavior. Front. Cell. Dev. Biol. 2021, 23, 745670. [Google Scholar] [CrossRef]
- Sikora, M.; Ermel, U.H.; Seybold, A.; Kunz, M.; Calloni, G.; Reitz, J.; Martin Vabulas, R.; Hummer, G.; Frangakis, A.S. Desmosome Architecture Derived from Molecular Dynamics Simulations and Cryo-Electron Tomography. Proc. Natl. Acad. Sci. USA 2020, 117, 27132–27140. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, A.P.; Green, K.J. Structure, Function, and Regulation of Desmosomes. Prog. Mol. Biol. Transl. Sci. 2013, 116, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Weis, W.I. Purification and Structural Analysis of Desmoplakin, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 569. [Google Scholar] [CrossRef]
- Choi, H.J.; Weis, W.I. Crystal Structure of a Rigid Four-Spectrin-Repeat Fragment of the Human Desmoplakin Plakin Domain. J. Mol. Biol. 2011, 409, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct from Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef]
- Yuan, Z.Y.; Cheng, L.T.; Wang, Z.F.; Wu, Y.Q. Desmoplakin and Clinical Manifestations of Desmoplakin Cardiomyopathy. Chin. Med. J. 2021, 134, 1771–1779. [Google Scholar] [CrossRef]
- Gomes, J.; Finlay, M.; Ahmed, A.K.; Ciaccio, E.J.; Asimaki, A.; Saffitz, J.E.; Quarta, G.; Nobles, M.; Syrris, P.; Chaubey, S.; et al. Electrophysiological Abnormalities Precede Overt Structural Changes in Arrhythmogenic Right Ventricular Cardiomyopathy Due to Mutations in Desmoplakin-A Combined Murine and Human Study. Eur. Heart J. 2012, 33, 1942–1953. [Google Scholar] [CrossRef]
- Daday, C.; Mateyka, L.M.; Gräter, F. How ARVC-Related Mutations Destabilize Desmoplakin: An MD Study. Biophys. J. 2019, 116, 831–835. [Google Scholar] [CrossRef]
- Stevens, T.L.; Manring, H.R.; Wallace, M.J.; Argall, A.; Dew, T.; Papaioannou, P.; Antwi-Boasiako, S.; Xu, X.; Campbell, S.G.; Akar, F.G.; et al. Humanized Dsp ACM Mouse Model Displays Stress-Induced Cardiac Electrical and Structural Phenotypes. Cells 2022, 11, 3049. [Google Scholar] [CrossRef]
- Hoover, C.A.; Ott, K.L.; Manring, H.R.; Dew, T.; Borzok, M.A.; Wright, N.T. Creating a ‘Molecular Band-Aid’; Blocking an Exposed Protease Target Site in Desmoplakin. J. Pers. Med. 2021, 11, 401. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G.; Spronk, C. YASARA–Yet Another Scientific Artificial Reality Application. YASARA.org 2013, 993, 51–78. [Google Scholar]
- Seeliger, D.; De Groot, B.L. Ligand Docking and Binding Site Analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G. Matrix Metalloproteinases: Regulation and Dysregulation in the Failing Heart. Circ. Res. 2002, 90, 520–530. [Google Scholar] [CrossRef]
- Vimalanathan, A.K.; Ehler, E.; Gehmlich, K. Genetics of and Pathogenic Mechanisms in Arrhythmogenic Right Ventricular Cardiomyopathy. Biophys. Rev. 2018, 10, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Julien, O.; Wells, J.A. Caspases and Their Substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Guo, J.; Zhang, X.; Sukhova, G.K.; Libby, P.; Shi, G.P. Cysteine Protease Cathepsins in Cardiovascular Disease: From Basic Research to Clinical Trials. Nat. Rev. Cardiol. 2018, 15, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Cieplak, P.; Strongin, A.Y. Matrix Metalloproteinases–From the Cleavage Data to the Prediction Tools and Beyond. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1952–1963. [Google Scholar] [CrossRef] [PubMed]
- Ratnikov, B.I.; Cieplak, P.; Gramatikoff, K.; Pierce, J.; Eroshkin, A.; Igarashi, Y.; Kazanov, M.; Sun, Q.; Godzik, A.; Osterman, A.; et al. Basis for Substrate Recognition and Distinction by Matrix Metalloproteinases. Proc. Natl. Acad. Sci. USA 2014, 111, E4148–E4155. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romov, I.M.; Nowzari, R.A.; Page, C.P.; Benes, M.R.; Borzok, M.A.; Wright, N.T. Prevention of Protease-Induced Degradation of Desmoplakin via Small Molecule Binding. J. Pers. Med. 2024, 14, 163. https://doi.org/10.3390/jpm14020163
Romov IM, Nowzari RA, Page CP, Benes MR, Borzok MA, Wright NT. Prevention of Protease-Induced Degradation of Desmoplakin via Small Molecule Binding. Journal of Personalized Medicine. 2024; 14(2):163. https://doi.org/10.3390/jpm14020163
Chicago/Turabian StyleRomov, Isabel M., Roujon A. Nowzari, Clay P. Page, Madeleine R. Benes, Maegen A. Borzok, and Nathan T. Wright. 2024. "Prevention of Protease-Induced Degradation of Desmoplakin via Small Molecule Binding" Journal of Personalized Medicine 14, no. 2: 163. https://doi.org/10.3390/jpm14020163