Abstract
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive systemic disease involving the extracellular deposition of misfolded transthyretin protein. The hereditary subtype is caused by mutations in the transthyretin (TTR) gene. An estimated 2–3% of individuals of African American (AA) ancestry carry the p.Val142Ile (V142I, also referred to as V122I) TTR pathogenic variant. The non-specific clinical nature of ATTR-CM makes it challenging to diagnose clinically, and the high allele frequency of TTR V142I suggests that many patients with hereditary ATTR-CM may not have been tested. An analysis of electronic health record data from over 13,000 AA patients with a diagnostic code for heart disease or arrhythmia who also had additional amyloid-related findings were not diagnosed with amyloidosis at higher rates than those with heart failure or arrhythmia who did not have additional amyloid-related clinical diagnoses. Similarly, after genotyping 666 AA patients with heart failure or arrhythmia, TTR V142I carriers appeared to be clinically indistinguishable based on amyloid-related non-cardiac diagnoses from those who did not carry the allele. No additional TTR gene sequence variants were found in the TTR wildtype V142V patients with heart failure or arrhythmia who had additional amyloid-related diagnoses. Genetic testing for ATTR-CM may be important for timely diagnosis.
1. Introduction
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive systemic disease involving the extracellular deposition of misfolded transthyretin protein [1]. Transthyretin is produced in the liver and circulates in the blood as a tetramer transporting thyroxin and retinol [2]. There are both mutation-related (hereditary) and wildtype (previously referred to as senile cardiac amyloidosis) forms of ATTR-CM [3]. Mutations in the transthyretin gene (TTR) increase tetramer dissociation, misfolding, and the aggregation of monomers into oligomeric amyloid fibrils, which can deposit in the heart and peripheral nervous system. The kinetics of monomer oligomerization appear to be related to amyloidogenicity, with disease-causing variants increasing the rates of aggregation. Increasing age is also associated with TTR tetramer instability.
The hereditary subtype is caused by autosomal dominantly acting mutations in the TTR [4]. More than 100 likely/pathogenic TTR variants have been described [5], with most mutations classified as single-nucleotide variants that cause substitutions of amino acids in the TTR protein monomer. There is extreme variability in the penetrance, expressivity, age of onset, and speed of progression, and whether neuropathy, cardiomyopathy, or other symptoms are the predominant phenotype is mutation-dependent. For example, the V30M variant can occur as an early-onset (<50 years of age) neurological disorder, which is found more frequently in Japan, Portugal, and Sweden than the later-onset disease, which also has cardiac manifestations. The T60A TTR variant that appears to have originated in Ireland also has both neurological- and cardiomyopathy-related symptoms. The allele frequencies of these variants are rare compared to p.Val142Ile (V142I, also referred to as V122I), carried by an estimated >3% of individuals of African American (AA) ancestry. V142I has been associated primarily but not exclusively with ATTR-CM [6]. A systematic literature review of the prevalence and outcomes of V142I ATTR-CM based on an aggregate sample size of ~150,000 found that the adjusted odds ratio for heart failure (HF) ranges from ~1.5 to 1.8 [7].
Despite the high prevalence of the TTR V142I variant in AA, there appears to be a substantial divergence between the number of TTR V142I carriers and diagnosed cases of ATTR-CM [8], suggesting that many patients, perhaps most, are undiagnosed, particularly those with heart failure who do not have overt clinical findings to suggest amyloidosis. In addition, ATTR-CM-related ventricular hypertrophy or heart failure may be difficult to distinguish from other prevalent causes of heart disease, such as hypertensive and hypertrophic cardiomyopathy, which are also highly prevalent [6]. Similarly, several types of arrhythmias, particularly atrial fibrillation, can occur with ATTR-CM [9] but are often ascribed to other causes. Some studies have reported non-cardiac manifestations of amyloidosis in TTR V142I carriers, including polyneuropathy and carpal tunnel syndrome, though the actual sample sizes of older patients were small [10]. The biological, genetic, and environmental factors that lead to the phenotypic penetrance and variable expressivity of ATTR-CM, thus, remain largely unknown.
Therapies have been developed to treat ATTR-CM, including antisense oligonucleotides, small interfering RNAs (ribonucleic acids), and CRISPR–Cas9 (clustered regularly interspaced short palindromic repeats-CRISPR-associated protein 9) gene editing, as well as small-molecule drugs [11]. Decreasing TTR protein translation in the liver to decrease circulating blood levels can produce corresponding reductions in amyloid deposition. Two primary approaches that have been developed and are now approved for clinical use are antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs). sASOs are short (~20 bases) single-strand oligonucleotides that are synthesized to hybridize to complementary sequences in the TTR mRNA. The ASO DNA-TTR mRNA hybrid is then targeted by the cell for degradation by ribonuclease RNase H1, depleting mRNA levels and reducing translation of the monomer protein. Similarly, siRNAs are slightly longer double-strand synthetic RNAs that also hybridize to the TTR mRNA, which is then degraded by the RNA-induced silencing complex (RISC). Although drugs from both of these classes have been approved for TTR-related polyneuropathy, siRNA has been associated with effectiveness against ATTR-CM in patients with mixed disease. The use of CRISPR–Cas9 to inactivate TTR in vivo is also being investigated. Small-molecule drugs to prevent TTR tetramers from dissociating have also been approved.
The non-specific nature of ATTR-CM makes it challenging to diagnose clinically, and the high allele frequency of TTR V142I suggests that many patients with hereditary ATTR-CM have not been thoroughly or formally evaluated and tested. With the growing repertoire of recently developed drugs to treat TTR-related amyloidosis [12], a significant health disparity exists for undiagnosed patients. We sought to determine whether carriers of TTR V142I of AA ancestry with heart failure or arrhythmia had extra-cardiac manifestations of amyloidosis, which could help suggest the diagnosis of amyloidosis. We studied patients seen in the Temple University Hospital System (TUHS), a catchment area with a high proportion of residents who self-identify as AA [13] and a high rate of cardiovascular disease. We also determined whether non-carriers of TTR V142I with heart failure or arrhythmia plus other non-cardiac manifestations of amyloidosis had other TTR variants.
2. Materials and Methods
2.1. Participants
Samples for DNA (deoxyribonucleic acid) isolation were obtained from clinically ordered EDTA anti-coagulated whole blood samples analyzed by the Temple University Health System (TUHS) Clinical Laboratory. The samples were collected from May 2021 to 2022 and stored for up to three days prior to biobanking. The Institutional Review Board (IRB) of Temple University approved the research. The electronic health record data of patients treated at Temple University Hospital, an urban academic medical center in Philadelphia, Pennsylvania, USA, were retrospectively analyzed using ICD-10 (International Classification of Diseases, Tenth Revision) codes (Table 1).

Table 1.
ICD-10 codes and corresponding diagnoses used in analysis of electronic health record data to identify patients with manifestations of ATTR-related amyloidosis.
2.2. Blood Samples and DNA Preparation
Genomic DNA for sequencing was isolated using the EZ1 DNA Blood 200 μL kit on the EZ1 Advanced XL instrument (Qiagen, Valencia, CA, USA). The DNA was extracted from EDTA anti-coagulated whole blood using the Qiagen MagAttract DNA Blood Midi M48 kit and the Qiagen BioRobot M48 Workstation (Qiagen) according to the manufacturer’s directions. Quantification of the extracted DNA was performed using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
2.3. TTR V142I Genotype Analysis
Genotyping for the p.Val142Ile (chr18:31598655, c.424G>A, rs76992529) variant was performed using a custom TaqMan genotyping assay that used probe fluorophores, VIC and FAM, to distinguish G from A on the Applied Biosystems QuantStudio 7 Flex System (Thermo Fisher Scientific Waltham, MA USA). The primer sequences were the forward primer CTGAGCCCCTACTCCTATTCCA, reverse primer GGAGGAGAAGTCCCTCATTCCTT, Probe 1 (VIC) ACGGCTGTCGTCACCAA, and Probe 2 (FAM) ACGGCTGTCATCACCAA, with the positions of the forward and reverse primers in the TTR gene sequence, as shown in Method S1. The DNA was genotyped according to the manufacturer’s protocol. The reaction components for each genotyping reaction were as follows: 10 ng of DNA, 5 μL of TaqMan Genotyping Master Mix (Applied Biosystems, Foster City, CA, USA), 0.25 μL of assay mix (40×), and water up to a total volume of 10 μL. The cycling conditions were 1 cycle at 60 °C for 30 s, followed by 1 cycle at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min, followed by 1 cycle at 60 °C for 30 s. The reaction was then analyzed by TaqMan Genotyper Software version 1.6.0.
2.4. TTR Gene Sequencing
The entire TTR gene was amplified on the Veriti Thermocycler as a 7792 bp fragment using long PCR (polymerase chain reaction) with the long PCR forward primer ACGAATGTTCCGATGCTCTAAT and reverse primer TGAGTTGCTGCAGGTGTATC. Following gel purification, the extracted DNA was verified by gel electrophoresis (Method S2), excised and purified, quantified, and then subjected to Sanger sequencing (Genewiz, Inc., South Plainfield, NJ USA) using 10 μL of purified PCR product, 2.5 μL of 10 μM sequencing primer, and 2.5 μL H2O. The sequencing primers (diagrammed in Method S2) were exon 1 forward primer ACGAATGTTCCGATGCTCTAAT and reverse primer AGTTCAAGTCCCAGCTCAGTAAG, exon 2 forward primer TGGGATCAGTGTGTAATTCTTGTTT and reverse primer CACAGCTAGAGGAGAGGAGTTCT, exon 3 forward primer AGGAGTTTTCCCTACTTCTGACTTA and reverse primer ATAGGAAAGGGAACCTTTGGTCATT, and exon 4 forward primer ACTTCCGGTGGTCAGTCATGTG and reverse primer TTAATACGTGCTTTGCTTGCAAGA. To identify the variants, FASTA sequence data were aligned to the reference genome using BLAST, and the fluorescent trace electrophoretograms were visually inspected.
2.5. Bioinformatics Analysis
Because the automated base calling of sequence data using the ABI 3730xl DNA Analyzer Sequencing Analysis KB Basecaller software cannot distinguish coincident base peaks characteristics of heterozygous variants, each Sanger sequencing capillary electrophoretogram was visually inspected to identify and confirm the variants. The NCBI BLAST tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 8 January 2024) was used to align the DNA sequence to the TTR sequence (NG_016441.1) and to translate the DNA sequence to its corresponding protein sequence. The NCBI gene database (https://www.ncbi.nlm.nih.gov/gene, accessed on 8 January 2024) was used to access TTR orthologs.
2.6. Statistical Analysis
All statistical tests were two-sided and conducted using Prizm 10.0, with p ≤ 0.05 considered statistically significant.
3. Results
Electronic health record data were extracted for over 13,000 TUHS AA patients with a diagnostic code for heart failure or arrhythmia as well as codes for other manifestations of ATTR-related amyloidosis, as described by expert consensus recommendations [14]. The ICD-10 codes used for data extraction are shown in Table 1.
Of the 13,029 AA patients with a diagnosis of either heart failure or arrhythmia, only 82 (0.63%) also had a diagnostic code for amyloidosis due to any cause (Table 2). Of the various amyloidosis diagnoses (Table S1), over half were coded as either “Organ-limited amyloidosis”, “Amyloidosis, unspecified”, or “Other amyloidosis”. The percentages of AA patients with a diagnosis of heart failure or arrhythmia and amyloidosis who also had non-cardiac amyloidosis findings varied by diagnosis. The percentages of patients with heart failure or arrhythmia and amyloidosis and no other diagnoses were not different from those who also had amyloidosis-related diagnoses (including spinal stenosis, spinal radiculopathy, bilateral carpal tunnel, orthostatic hypotension, bladder dysfunction, multi/chronic diarrhea, numbness or tingling, eye disease, spinal stenosis or spinal radiculopathy and eye diseases, multi/chronic diarrhea and spinal stenosis, multi/chronic diarrhea, and spinal radiculopathy). Only the percentage of patients with heart failure or arrhythmia and gastroparesis was nominally different (p = 0.029, Fisher’s exact test). However, this result was not significant after correction for multiple testing. In contrast, the percentage of patients with heart failure or arrhythmia and amyloidosis and left ventricular hypertrophy was over four-fold higher (nominal p = 0.015, Fisher’s exact test) than that of those with heart failure or arrhythmia and amyloidosis without left ventricular hypertrophy. Similarly, the percentage of patients with heart failure or arrhythmia and amyloidosis was over eight-fold higher in those who also had an atrioventricular block (p = 0.022, Fisher’s exact test).
Table 2.
Non-cardiac amyloidosis diagnoses in TUHS AA patients with heart failure (HF) or arrhythmia.
To estimate the prevalence of TTR V142I, the DNA obtained from the blood samples of 666 TUHS AA patients with a diagnosis of heart failure or arrhythmia included in a biobank of samples was genotyped. The TaqMan genotyping (Figure 1A) assay was an efficient and robust method to identify V142I. Approximately 2.7% of the patients carried a TTR V142I allele (18/666; 1 homozygote, 17 heterozygotes). The TTR V142I homozygote was >65 years and female, with a diagnosis of HF or arrhythmia plus spinal stenosis, bilateral carpal tunnel syndrome, multi/chronic diarrhea, eye disease, and lower extremity numbness. Of the 18 TTR V142I allele carriers, 38.9% were male, and 50% were 65 years of age or older. This percentage of males was not different from those <65 years of age (p < 0.475, Fisher’s exact test). Similar percentages for age and sex distribution were found for the V142V reference sequence individuals. Although the percentages of most of the non-cardiac findings were higher in the TTR V142I carriers, none were statistically significant (Table 3 and Figure 2).
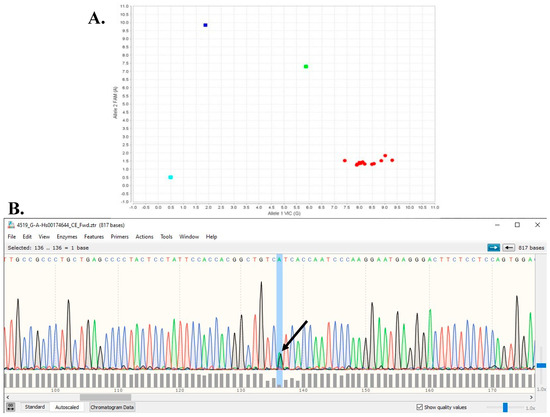
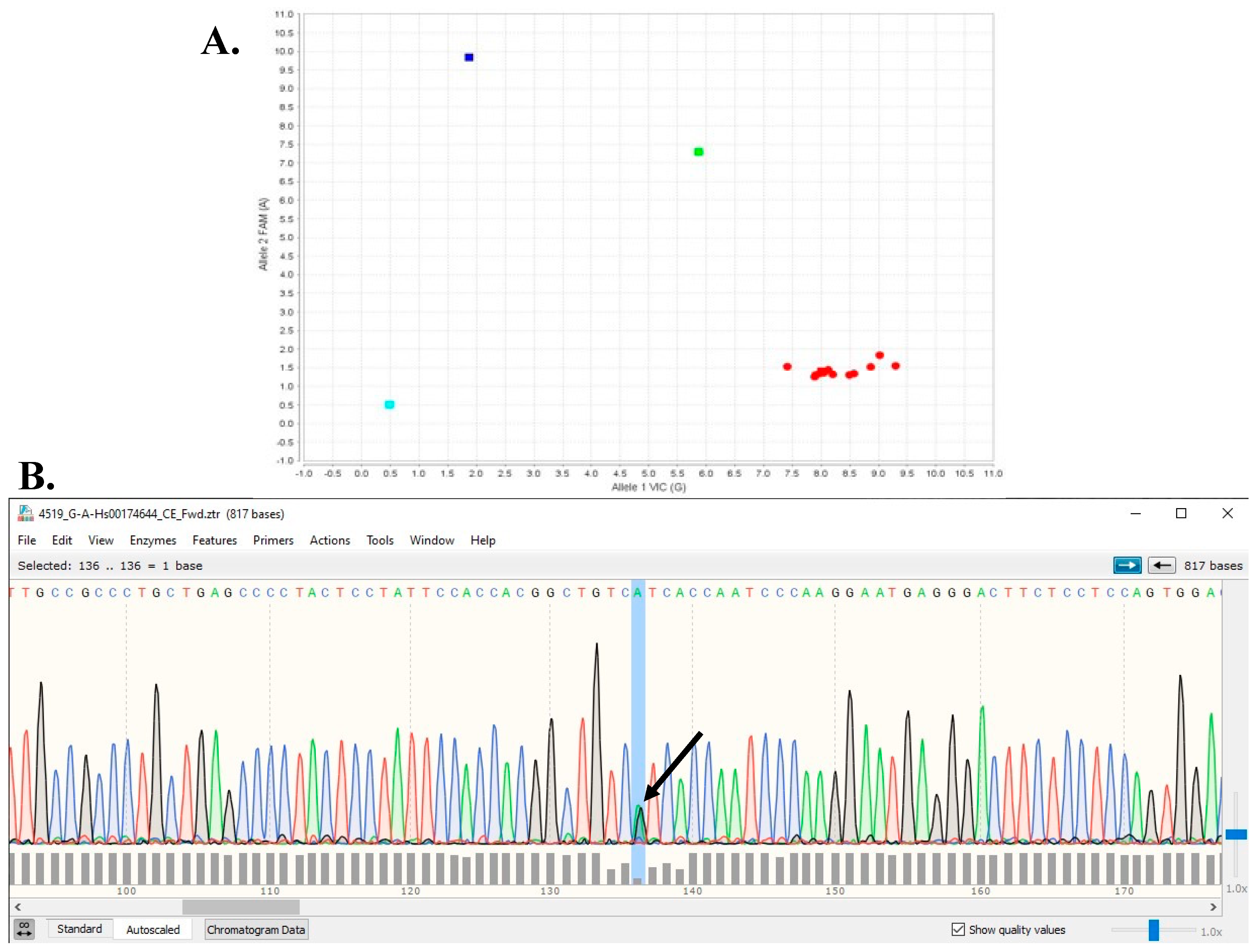
Figure 1.
(A) Example of TTR Taqman genotyping assay result for TTR V142I. Blue square is homozygous V142I/V142I, green square is heterozygous V142V/V142I, and red squares are homozygous reference V142V/V142V (turquoise square is baseline negative control for assay). (B) Example of Sanger sequencing chromatogram. Each fluorescence tracing peak color corresponds to each of the four DNA bases, i.e., green = A, red = T, black = G, and blue = C peaks Arrow indicates overlapping shorter A (green) and G (black) peaks for heterozygous V142V/V142I.
Table 3.
Percentages of patients with or without the p.Val142Ile variant by diagnostic category.
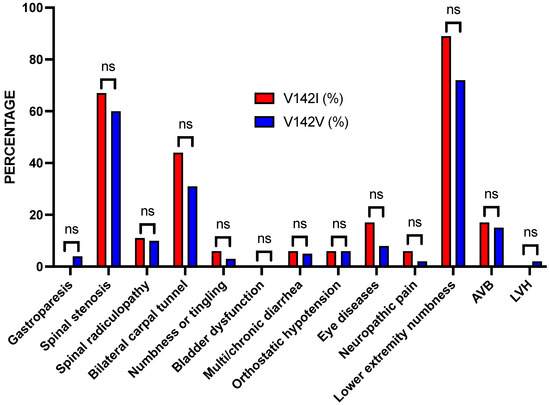
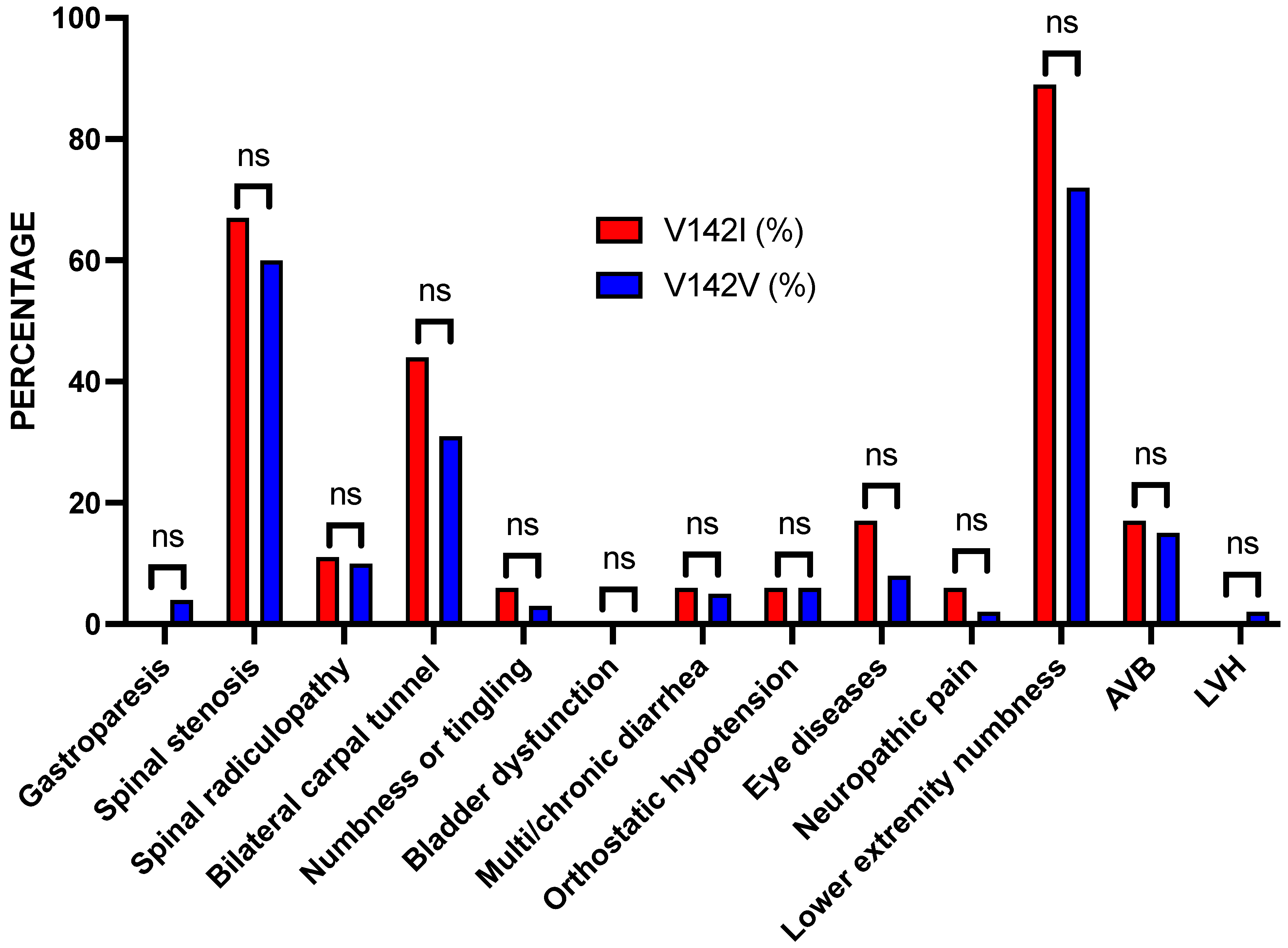
Figure 2.
Bar graph of the percentages of patients with the TTR V142I variant or the reference V142V sequence by diagnostic category. Although the percentages of most of the non-cardiac findings were higher in the TTR V142I carriers, all were not significant (ns).
We then sequenced (Figure 1B) the 27 patients with a diagnosis of heart failure or arrhythmia who did not carry TTR V142I, i.e., they had the TTR V142V wildtype but also had other amyloidosis-related diagnoses (Table S1). All 27 had bilateral carpal tunnel, 25 had lower extremities numbness, 23 had spinal radiculopathy, and 21 had spinal stenosis. The number of amyloidogenic diagnoses ranged from 2 to 8, with a median of 5. Variants were identified in 23/27 patients (Table S2). A total of 22/27 patients had intronic variants, 3 patients had variants in the 3′ untranslated coding region, and no missense or other exon variants were found in any of the patients. Accessing the gnomAD (genome Aggregation Database) database [15] revealed that of the 23 TTR missense variants, only 4 other than V142I were present in the African American population: p.Ile88Leu, p.Thr80Ile, p.Val50Met, and p.Thr126Asn (Table S3). The allele frequency in AA for these four were 4/74,294, 1/59,116, 1/75,034, and 1/41,466, respectively. In contrast, in the European non-Finnish population, the respective allele frequencies for the four variants were 17/1,179,982, 0/418,132, 62/1,179,988, and 0/68,038. Although there is no information on any clinical conditions in individuals in gnomAD, given the later age of onset and variable penetrance and expressivity of ATTR-CM, these data suggest that apart from V142I, TTR variants are rare in AA. Valine at position 142 is also highly conserved, present in about 90% of species in the NCBI (National Center for Biotechnology Information) Gene database, with TTR orthologs.
4. Discussion
The data from the TUHS indicate that approximately 0.63% of AA patients with a diagnosis of either heart failure or arrhythmia also carry a diagnosis of amyloidosis. In patients with either heart failure or arrhythmia, plus the diagnosis of gastroparesis, bilateral carpal tunnel, orthostatic hypotension, or chronic diarrhea, the percentage of patients with a diagnosis of amyloidosis increased by 2–4-fold, though none of the increases met statistical significance after correcting for multiple testing. Similarly, after genotyping 666 AA patients with a diagnosis of either heart failure or arrhythmia, the percentages of V142I and V142V patients did not have any significant differences in the diagnoses of 11 non-cardiac amyloid-related diagnoses and 2 cardiac diagnoses. These data further suggest that TTR V142I in AA predominantly manifests as a cardiac phenotype related to heart failure [16,17,18], supported by a large systematic review of the literature [7]. The data showing the contrary have been largely based on very small sample sizes [19].
We found no other TTR variants in AA patients with either heart failure or arrhythmia who also had multiple other diagnoses associated with TTR amyloidosis. Given the relatively cardiac-specific phenotype of TTR V142I ATTR-CM, analyzing a small sample size of heart failure patients with additional amyloid diagnoses likely did not enrich for patients with TTR mutations. In addition, based on the gnomAD database, the frequency of non-V142I TTR mutations is very low.
Why V142I has a high allele frequency for a dominantly acting pathogenic variant is not clear. Humans who lack TTR do not appear to have been described [20]; thus, the gene likely has some essential function. However, mice without TTR, i.e., TTR knockouts, appeared to be phenotypically unaffected, with normal viability and fertility [21]. The initial designation of TTR as prealbumin, because its migration was ahead of albumin in serum protein electrophoresis, was changed to thyroxine-binding prealbumin following the discovery that it transported thyroid hormone [22]. After it was also found to bind to retinol-binding protein, transthyretin was found to transport (trans-) thyroid hormone (-thyr-) and retinol-binding protein (-retin). The valine at position 142 is highly conserved, suggesting that this particular amino acid is important for TTR function. The substitution of isoleucine for valine appears to destabilize TTR tetramers [23]. The 142 valine position appears to be in a conserved β-sheet in strand H of the TTR protein, which corresponds to a serine in a highly conserved YRGS motif that is present in non-vertebrate TTR homologs but absent from vertebrates [24]. The function of this YRGS motif is not known.
How the V142I variant predisposes someone to cardiac manifestations in favor of neurological ones is not clear. The V142I variant appears to destabilize the TTR homotetramer relative to the reference V142V homotetramer [25]. Amino acid 142 is on the periphery of the H strand β-sheet, which forms part of the quaternary structural interface of the homotetramer. Antiparallel β-sheet interactions occur between the H strands of two monomers, contributing to the stability of the dimer interface. The side chain of the mutant 142 isoleucine has altered interactions (relative to the valine at 142) with the side chains of the amino acids of the neighboring subunit. The altered interactions at the dimer–dimer interface cause the tetramer instability. The V142I TTR monomer that results can undergo a rapid partial denaturation, which allows for self-assembly into amyloid fibrils. The tissue specificity of the amyloid deposition may be related to the physico-chemical characteristics of the resulting fibrils [26].
With the relatively high allele frequency of V142I and the estimated adjusted odds ratio for the risk of heart failure estimated at ~1.5–1.8 in TTR V142I carriers [7], many patients are either undiagnosed or at risk for being diagnosed in the future. In a study of 278 AA individuals who were ≥60 years old with heart failure, 19 (6.8%) had positive cardiac technetium-99m-pyrophosphate imaging indicative of amyloid, with 7 (37%) of these individuals genotyping as carriers of V142I [8]. These data further support the likely underdiagnosis of ATTR-CM due to TTR V142I.
Currently, the main diagnostic algorithm for patients with suspected cardiac amyloidosis utilizes genetic testing as a near-final step. Diagnostic modalities recommended prior to genetic analysis include electrocardiography, echocardiography, cardiac magnetic resonance imaging, biomarkers, serum kappa/lambda free light chain ratio, serum and urine immunofixation, and endomyocardial biopsy and/or myocardial scintigraphy with bone avid tracers [27,28]. Although these methods can detect manifestations of ATTR-CM, the wide array of clinical signs and symptoms and heterogeneity of the cardiac phenotype can result in diagnostic delays or misdiagnosis [29]. Genetic testing as an almost final diagnostic step may also be overlooked, compounded by the prevailing view that genetic testing is expensive. However, genetic testing has now become low-cost (and for TTR amyloidosis can be free), non-invasive, and is now widely used in medicine [30]. The use of genetic testing would both streamline the diagnostic process and improve the rates of accurate diagnosis. This may be particularly important for V142I ATTR-CM, which may manifest a more aggressive cardiac phenotype and poorer prognosis [31] and an increase in all-cause mortality relative to V142I noncarriers [17].
Several TTR-directed treatments are now available for ATTR-CM. However, symptomatic therapy with heart failure medications is commonly used, particularly prior to the diagnosis. The clinical benefit of guideline-directed therapy for patients with HFrEF (heart failure with reduced ejection fraction) in ATTR-CM, including beta-blockers and angiotensin-converting enzyme inhibitors, is not clear [32]. Other drugs, such as calcium channel blockers and digitalis, may be contraindicated [33]. In addition, delayed diagnosis will result in the delayed initiation of appropriate treatment. ATTR-CM patients who experienced delays in diagnosis and treatment were found to have poor prognostic indicators, including a higher NYHA (New York Heart Association) classification, increased cardiac biomarkers, and lower health-related quality of life [34]. For this reason, using genetic testing to diagnose ATTR-CM in heart failure or arrhythmia in AA patients with or without additional clinical indications may be important for the timely initiation of appropriate therapy.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jpm14030271/s1, Table S1. Non-cardiac clinical manifestations; Table S2. Sanger sequencing variants; Table S3. gnomAD TTR variants; Table S4. AA missense TTR variants in gnomAD (v4.0.0) by population; Method S1. Genotyping; Method S2. Sanger sequencing.
Author Contributions
Conceptualization, G.S.G.; methodology, G.S.G., E.H., C.F. and S.K.; formal analysis, G.S.G.; investigation, S.M.O., L.I., F.C., D.L., N.N., L.S. and Y.V.; data curation, S.K.; writing—original draft preparation, G.S.G., S.K. and L.I.; writing—review and editing, G.S.G., E.H., C.F., S.K. and L.I.; project administration, G.S.G. and S.K.; funding acquisition, G.S.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by funds from Pfizer grant #57279519 and Genentech G-76756.
Institutional Review Board Statement
This study was approved by the Institutional Review Board of Temple University, #25673, approved 14 February 2019.
Informed Consent Statement
Patient consent was waived because all samples and data were de-identified prior to analysis for scientific purposes.
Data Availability Statement
All data from this study are presented in the main text or Supplemental Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Saito, Y.; Nakamura, K.; Ito, H. Molecular Mechanisms of Cardiac Amyloidosis. Int. J. Mol. Sci. 2021, 23, 25. [Google Scholar] [CrossRef]
- Zegkos, T.; Gossios, T.; Ntelios, D.; Parcharidou, D.; Karvounis, H.; Efthimiadis, G. Wild-Type Transthyretin Amyloid Cardiomyopathy: The Gordian-Knot of Novel Therapeutic Regimens. Cardiol. Rev. 2023, 31, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Dispenzieri, A.; Sher, T. Pathophysiology and treatment of cardiac amyloidosis. Nat. Rev. Cardiol. 2015, 12, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Mankad, A.K.; Shah, K.B. Transthyretin Cardiac Amyloidosis. Curr. Cardiol. Rep. 2017, 19, 97. [Google Scholar] [CrossRef]
- Bukhari, S. Cardiac amyloidosis: State-of-the-art review. J. Geriatr. Cardiol. 2023, 20, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.N.; Ruberg, F.L. Transthyretin V122I (pV142I)* cardiac amyloidosis: An age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet. Med. 2017, 19, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, P.; Alhuneafat, L.; Mannello, M.; Al-Rashdan, L.; Kim, M.M.; Dungu, J.; Alexander, K.; Masri, A. Prevalence and Outcomes of p.Val142Ile TTR Amyloidosis Cardiomyopathy: A Systematic Review. Circ. Genom. Precis. Med. 2021, 14, e003356. [Google Scholar] [CrossRef]
- Madhani, A.; Sabogal, N.; Massillon, D.; Paul, L.D.; Rodriguez, C.; Fine, D.; Helmke, S.; Winburn, M.; Kurian, D.; Raiszadeh, F.; et al. Clinical Penetrance of the Transthyretin V122I Variant in Older Black Patients with Heart Failure: The SCAN-MP (Screening for Cardiac Amyloidosis with Nuclear Imaging in Minority Populations) Study. J. Am. Heart Assoc. 2023, 12, e028973. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Lo, P.; Cho, K.; Subbiah, R. Ventricular Arrhythmias in Cardiac Amyloidosis: A Review of Current Literature. Clin. Med. Insights Cardiol. 2020, 14, 1179546820963055. [Google Scholar] [CrossRef]
- Parker, M.M.; Damrauer, S.M.; Tcheandjieu, C.; Erbe, D.; Aldinc, E.; Hawkins, P.N.; Gillmore, J.D.; Hull, L.E.; Lynch, J.A.; Joseph, J.; et al. Association of the transthyretin variant V122I with polyneuropathy among individuals of African ancestry. Sci. Rep. 2021, 11, 11645. [Google Scholar] [CrossRef]
- Aimo, A.; Castiglione, V.; Rapezzi, C.; Franzini, M.; Panichella, G.; Vergaro, G.; Gillmore, J.; Fontana, M.; Passino, C.; Emdin, M. RNA-targeting and gene editing therapies for transthyretin amyloidosis. Nat. Rev. Cardiol. 2022, 19, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Tomasoni, D.; Bonfioli, G.B.; Aimo, A.; Adamo, M.; Canepa, M.; Inciardi, R.M.; Lombardi, C.M.; Nardi, M.; Pagnesi, M.; Riccardi, M.; et al. Treating amyloid transthyretin cardiomyopathy: Lessons learned from clinical trials. Front. Cardiovasc. Med. 2023, 10, 1154594. [Google Scholar] [CrossRef] [PubMed]
- Le Guen, C.L.; King, N.A.; Zhao, H.; Renza-Stingone, E.P.; Gerhard, G.S.; Soans, R.S. COVID-19 patients with obesity at risk for worse outcomes despite younger age and fewer inflammatory derangements. Surg. Obes. Relat. Dis. 2021, 17, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Ando, Y.; Beirao, J.M.; Coelho, T.; Gertz, M.A.; Gillmore, J.D.; Hawkins, P.N.; Lousada, I.; Suhr, O.B.; Merlini, G. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J. Neurol. 2021, 268, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Author Correction: The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2021, 590, E53. [Google Scholar] [CrossRef] [PubMed]
- Parcha, V.; Malla, G.; Irvin, M.R.; Armstrong, N.D.; Judd, S.E.; Lange, L.A.; Maurer, M.S.; Levitan, E.B.; Goyal, P.; Arora, G.; et al. Association of Transthyretin Val122Ile Variant with Incident Heart Failure Among Black Individuals. JAMA 2022, 327, 1368–1378. [Google Scholar] [CrossRef]
- Kozlitina, J.; Garg, S.; Drazner, M.H.; Matulevicius, S.A.; Ayers, C.; Overton, J.; Reid, J.; Baras, A.; Rao, K.; Pandey, A.; et al. Clinical Implications of the Amyloidogenic V122I Transthyretin Variant in the General Population. J. Card. Fail. 2022, 28, 403–414. [Google Scholar] [CrossRef]
- Damrauer, S.M.; Chaudhary, K.; Cho, J.H.; Liang, L.W.; Argulian, E.; Chan, L.; Dobbyn, A.; Guerraty, M.A.; Judy, R.; Kay, J.; et al. Association of the V122I Hereditary Transthyretin Amyloidosis Genetic Variant with Heart Failure Among Individuals of African or Hispanic/Latino Ancestry. JAMA 2019, 322, 2191–2202. [Google Scholar] [CrossRef]
- Shije, J.Z.; Bautista, M.A.B.; Smotherman, C. The Frequency of V122I Transthyretin Mutation in a Cohort of African American Individuals With Bilateral Carpal Tunnel Syndrome. Front. Neurol. 2022, 13, 949401. [Google Scholar] [CrossRef]
- Liz, M.A.; Coelho, T.; Bellotti, V.; Fernandez-Arias, M.I.; Mallaina, P.; Obici, L. A Narrative Review of the Role of Transthyretin in Health and Disease. Neurol. Ther. 2020, 9, 395–402. [Google Scholar] [CrossRef]
- Episkopou, V.; Maeda, S.; Nishiguchi, S.; Shimada, K.; Gaitanaris, G.A.; Gottesman, M.E.; Robertson, E.J. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc. Natl. Acad. Sci. USA 1993, 90, 2375–2379. [Google Scholar] [CrossRef]
- McLean, T.R.; Rank, M.M.; Smooker, P.M.; Richardson, S.J. Evolution of thyroid hormone distributor proteins. Mol. Cell Endocrinol. 2017, 459, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Hurshman Babbes, A.R.; Powers, E.T.; Kelly, J.W. Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: The relationship between stability and amyloidosis. Biochemistry 2008, 47, 6969–6984. [Google Scholar] [CrossRef] [PubMed]
- Hennebry, S.C.; Wright, H.M.; Likic, V.A.; Richardson, S.J. Structural and functional evolution of transthyretin and transthyretin-like proteins. Proteins 2006, 64, 1024–1045. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Buxbaum, J.N.; Kelly, J.W. The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc. Natl. Acad. Sci. USA 2001, 98, 14943–14948. [Google Scholar] [CrossRef]
- Poli, L.; Labella, B.; Cotti Piccinelli, S.; Caria, F.; Risi, B.; Damioli, S.; Padovani, A.; Filosto, M. Hereditary transthyretin amyloidosis: A comprehensive review with a focus on peripheral neuropathy. Front. Neurol. 2023, 14, 1242815. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Bokhari, S.; Damy, T.; Dorbala, S.; Drachman, B.M.; Fontana, M.; Grogan, M.; Kristen, A.V.; Lousada, I.; Nativi-Nicolau, J.; et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Cardiac ATTR Amyloidosis. Circ. Heart Fail. 2019, 12, e006075. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.S.; Hawkins, P.N. Cardiac amyloidosis: Where are we today? J. Intern. Med. 2015, 278, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Elliott, P.; Comenzo, R.; Semigran, M.; Rapezzi, C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation 2017, 135, 1357–1377. [Google Scholar] [CrossRef]
- Semsarian, C.; Ingles, J.; Ross, S.B.; Dunwoodie, S.L.; Bagnall, R.D.; Kovacic, J.C. Precision Medicine in Cardiovascular Disease: Genetics and Impact on Phenotypes: JACC Focus Seminar 1/5. J. Am. Coll. Cardiol. 2021, 77, 2517–2530. [Google Scholar] [CrossRef]
- Razvi, Y.; Ioannou, A.; Patel, R.K.; Chacko, L.; Karia, N.; Riefolo, M.; Porcari, A.; Rauf, M.U.; Starr, N.; Ganesananthan, S.; et al. Deep phenotyping of p.(V142I)-associated variant ATTR amyloid cardiomyopathy: Distinct from wild-type ATTR amyloidosis? Eur. J. Heart Fail. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Kristen, A.V. Amyloid cardiomyopathy. Herz 2020, 45, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Elsanhoury, A. Treatment of Transthyretin Amyloid Cardiomyopathy: The Current Options, the Future, and the Challenges. J. Clin. Med. 2022, 11, 2148. [Google Scholar] [CrossRef] [PubMed]
- Rozenbaum, M.H.; Large, S.; Bhambri, R.; Stewart, M.; Whelan, J.; van Doornewaard, A.; Dasgupta, N.; Masri, A.; Nativi-Nicolau, J. Impact of Delayed Diagnosis and Misdiagnosis for Patients with Transthyretin Amyloid Cardiomyopathy (ATTR-CM): A Targeted Literature Review. Cardiol. Ther. 2021, 10, 141–159. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).