Abstract
Background/Objectives: BRCA1, BRCA2, ATM, and CHEK2 are known cancer predisposition genes (CPGs), but tumor risk in patients with simultaneous pathogenic variants (PVs) in CPGs remains largely unknown. In this study, we describe six patients from five families with multiple cancers who coinherited a combination of PVs in these genes. Methods: PVs were identified using NGS DNA sequencing and were confirmed by Sanger. Results: Families 1, 2, and 3 presented PVs in BRCA2 and ATM, family 4 in BRCA2 and BRCA1, and family 5 in BRCA2 and CHEK2. PVs were identified using NGS DNA sequencing and were confirmed by Sanger. The first family included patients with kidney, prostate, and breast cancer, in addition to pancreatic adenocarcinomas. In the second family, a female had breast cancer, while a male from the third family had prostate, gastric, and pancreatic cancer. The fourth family included a male with pancreatic cancer, and the fifth family a female with breast cancer. Conclusions: The early age of diagnosis and the development of multiple cancers in the reported patients indicate a very high risk of cancer in double-heterozygous patients associated with PVs in HR-related CPGs. Therefore, in families with patients who differ from other family members in terms of phenotype, age of diagnosis, or type of cancer, the cascade testing needs to include the study of other CPGs.
1. Introduction
Cancer predisposition syndromes (CPS) are now extensively studied, with an increasing proportion of cancer patients undergoing genetic testing [1]. This testing is based on the type of cancer, the number of cancer occurrences during the patient’s life, the age at diagnosis, and the family history [2,3]. It is expected that 3 to 5% of cancers are linked to a causal variant in a cancer predisposition gene (CPG) [4]. As most CPS are transmitted in an autosomal dominant way, once a pathogenic variant (PV) is identified in a family, the geneticists propose a family cascade testing to search for the variant, and start with first-degree relatives [5].
Within the frame of inherited cancer predisposition, carriers of pathogenic variants (PVs) in a single gene have been extensively represented in the literature, and an ever-growing accumulation of data on the single gene-related cancer risk, based on multiple family histories, is available [6,7]. These data have led to gene-specific screening and follow-up recommendations for these carriers [3]. However, the coinheritance of heterozygous PV in two CPGs is a poorly studied event restricted to small case series and single case reports [8,9,10]. The exact frequency of double heterozygotes remains unknown, as is the case for their cancer risk and associated follow-up strategies [11]. Therefore, empirically, most genetic centers propose to apply the guidelines defined for the most dangerous gene to the follow-up of patients with two PVs in two different CPGs. However, the BRCA1 and BRCA2 PVs coinheritance, in the population-based Israeli national breast cancer cohort, was described in 2.2% of all carriers [12], and 17 double heterozygotes for CPGs were detected in a breast cancer cohort of people of Slavic ancestry which included 5391 patients [13].
Breast cancer gene 1 (BRCA1), breast cancer gene 2 (BRCA2), checkpoint kinase 2 (CHEK2), and ataxia-telangiectasia mutated (ATM) are CPGs, part of the homologous recombination (HR) pathway for double strand break (DSB) repair. This pathway preferentially uses the sister chromatid for error-free repair, and both the DNA damage response and the cell cycle checkpoints are crucial for initiating and regulating HR [14]. ATM participates in HR initiation and phosphorylation of CHEK2; BRCA1 facilitates DNA end resection [15], while BRCA2 aids in the formation of a DNA D-loop through the invasion of the nearby duplex DNA [16]. Finally, the BRCA2 protein is post-translationally modified by ATM [17].
HR is crucial for repairing severe replication lesions at replication forks, and can repair or bypass DNA lesions remaining due to inactivation of other pathways. Consequently, mutations in HR genes result in genomic instability, fueling further mutations that lead to cancer development [14,18]. This deficiency in the HR pathway makes tumor cells more sensible to poly-(ADP-ribose)-polymerase inhibitors, platinum derivatives, alkylating agents, mitomycin C, and other antitumor drugs that are used for the treatment of cancer patients [19,20,21,22].
PVs in BRCA1, BRCA2, CHEK2, and ATM have been linked to a wide variety of cancers [15]. BRCA1 and BRCA2’s PVs were associated with breast cancer, ovarian/fallopian cancer, pancreas cancer, prostate cancer, and melanoma, while breast, prostate, thyroid, kidney, colon and stomach cancers were related to PVs in CHEK2 [23]. Germline heterozygous PVs in ATM increase the risks of breast, pancreatic, gastro-esophageal, colorectal, ovarian, prostate, thyroid, gastric, and head and neck cancers, as well as melanoma [24]. Given the frequencies of PVs in these genes, it is expected that cancer patients carrying two PVs should be rarely, but not exceptionally, observed. Moreover, as these genes act on the homologous recombination pathway, these double heterozygote patients might have a higher risk of HR dysfunction and thus a more severe cancer risk.
In this study we describe six patients from five families with multiple cancers who coinherited PVs in BRCA2 and other HR genes—four patients with variants in BRCA2 and ATM, one patient with BRCA2 and BRCA1, and one patient with BRCA2 and CHEK2 PVs.
2. Materials and Methods
2.1. Ethical Approval
The study was conducted in accordance with the Declaration of Helsinki and approved by the Comité d’Ethique Hospitalo-facultaire Universitaire de Liège (protocol code 2019/245 and date of approval 28 October 2019).
2.2. Data Collection
Patient sex, age, age at diagnosis for each tumor, and personal and family history were extracted from the medical records. Data on cancer diagnosis and treatment were gathered from the institution’s database. All of the patients read and signed an informed-consent document.
2.3. Genetic Analysis
Genetic analysis was performed on DNA extracted from blood samples using QIAcube (QIAGEN, Hilden, Germany) and STARlet (Seegene Inc., Seoul, South Korea) extraction instruments (See Supplementary Data: DNA extraction methods). DNA purity and concentration were measured with NanoDrop (Thermo Fisher Scientific, Waltham, Massachusetts, United States), and DNA underwent NGS panel sequencing (See Supplementary Data: Table S1). The bioinformatic analysis was performed using in-house demultiplexing pipelines and the in-house Humanomics pipeline (as described in [25]). Variant classification was performed according to the ACMG “Standards and guidelines for the interpretation of sequence variants” [26]. The in silico analysis of missense and splicing variants was performed using the aggregated score of the Franklin by Genoox tool (https://franklin.genoox.com, accessed on 17 May 2024), which includes the scores of SIFT, FATHMM, DANN, MetaLR, REVEL, MutationAssessor, PolyPhen-2, MutationTaster, PrimateAI, BayesDel, SpliceAI, dbscSNV, GERP, GenoCanyon, fitCons, MitoTip, and APOGEE. For the splicing variants, Human Splicing Finder [27] was used. Two databases, gnomAD (https://gnomad.broadinstitute.org/, accessed on accessed on 17 May 2024) and ALFA (https://www.ncbi.nlm.nih.gov/snp/docs/gsr/alfa, accessed on accessed on 17 May 2024), were used to retrieve the Minor Allele Frequency (MAF) data. The identified PVs were confirmed by Sanger sequencing (See Supplementary Materials: Table S2).
3. Results
3.1. Frequency of Double Heterozygotes
Over the past 28 months, following the introduction of the new Hereditary Breast and Ovarian Cancer (HBOC) panel at our institution, a total of 2152 panels have been conducted in cases of cancer patients (1929 13-gene panels and 223 26-gene panels). In total, 121/1929 13-gene panels (6.27%) and 22/223 (9.8%) 26-gene panels were positive, containing a pathogenic or likely pathogenic result. Three patients (3/2152 patients, 0.14%) were double-heterozygous for CPG PVs. Two samples had two PVs in the 13-gene panel (2.2% of the 91 samples with PVs) and one in the 26-gene panel (5.6% of the 18 samples with PVs, see Table 1). Heterozygous variants in genes associated with a recessive instance of CPS, such as the MUTYH gene, were excluded from this analysis.

Table 1.
Double heterozygote statistics in the institution.
In this study, we report two of the three double-heterozygous patients from whom we obtained informed consent, and one additional patient whose double-heterozygous state was diagnosed based on family history. The three additional included patients were previously observed by the genetics department and/or had a relevant family history.
3.2. Clinical History
Six patients from five families underwent genetic consultation in the context of multiple cancers or early-onset disease, leading to the identification of two heterozygous PVs in the HR genes of each patient (see Table 2).
Table 2.
Characteristics of the patients included in the study.
The first family included a 67-year-old male with a medical history of multiple cancers whose daughter had been diagnosed with breast cancer (see Figure 1). The male patient presented kidney and prostate cancer and pancreatic adenocarcinoma at the ages of 50, 51, and 66, respectively.
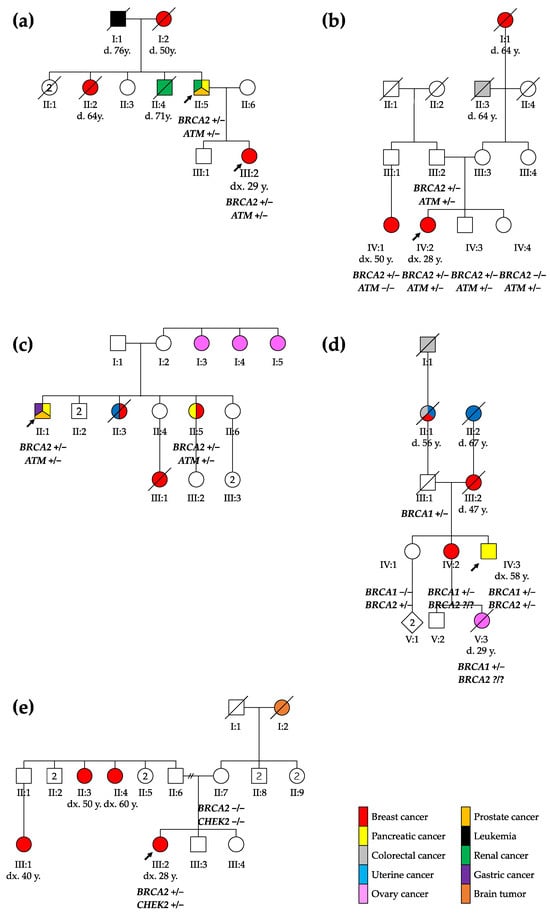
Figure 1.
Pedigree of the five families reported: (a) family 1, (b) family 2, (c) family 3, (d) family 4, and (e) family 5. The probands are marked with arrows. ca., cancer; CRC, colorectal cancer; d., death; dx., diagnosis; y., years; +/−, heterozygous genotype; −/−, homozygous wild type genotype; ?/?, unknown genotype.
The patient’s daughter was diagnosed with breast cancer at 29 years. A tumorectomy showed grade 3 invasive ductal carcinoma with axillary and mediastinal lymph node extension (ypT1cN2aMx). Two years after the diagnosis, she presented a first relapse with one successfully treated bone metastasis. The subsequent relapses included liver metastasis, lymph node invasion, and finally brain metastasis in 2021.
In the second family, a 28-year-old female underwent an exploratory laparoscopy due to persistent non-specific abdominal pain with nausea and vomiting, showing endometriotic lesions and multiple hepatic lesions described as angiomas. A month later, after a week of hyperthermy and a positive COVID-19 test, the thoracoabdominal computed tomography scan demonstrated a large breast lesion with a highly suspicious right axillary lymph node, necrotic hepatic and bone lesions, and possibly-COVID-19-related pulmonary foci. A grade 3 infiltrating ductal carcinoma was diagnosed and treated.
A 65-year-old male from the third family was diagnosed with a Gleason 3 pT2bNxM0 prostate adenocarcinoma at the age of 49, well-differentiated pT1N0M0 enteric adenocarcinoma one year later, and finally metastatic pancreatic cancer. His older sister was first diagnosed with breast cancer at 60, and then pancreatic cancer at 70 years old.
The fourth family included a 58-year-old male who presented a 15 kg weight loss, fatigue, nausea, and transfixing abdominal pain for 2 weeks. In a tomography, an isthmus pancreatic mass of 4 cm infiltrating peripancreatic fat with hepatic metastasis was discovered (CTxNxM1). In this patient, a familial BRCA1 variant was found 15 years earlier at the time of a breast cancer diagnosis for his sister at the age of 35 (she developed a second breast cancer 15 years later, and pancreatic cancer at the age of 67). The male patient was known to carry this familial BRCA1 variant, inherited from their father. As the BRCA1 familial variant was not sufficient to explain both pancreatic cancers, those in the patient and his sister, as well as their mother’s breast cancer, we re-initiated a CPG analysis and this showed that he carried two pathogenic variants: the known familial BRCA1 PV, and a BRCA2 PV.
The 29-year-old female from the fifth family discovered three mobile, not painful masses in her right breast while performing self-palpation. The biopsy of one of the masses revealed a ductal breast adenocarcinoma (cT2N0M0). After a right mastectomy with sentinel ganglion, an infiltrating tubular adenocarcinoma (pT2mN1mi) was diagnosed. During genetic evaluation, a BRCA2 and a CHEK2 PV were identified in the patient. Both PVs were absent in the mother, while the father was not available for testing. The patient has two sisters, one of whom is underage and has not been tested.
3.3. Genetic Characteristics
In the patients from families 1 to 3, genetic analyses showed BRCA2 and ATM PVs. The patients from family 4 and 5 carried PVs in BRCA1/BRCA2 and BRCA2/CHEK2, respectively.
Three of the identified BRCA2 nonsense variants were located in exon 11/27 (c.3865_3868del, c.5057T>A, c.4284dup), while the fourth was located in exon 7/27 (c.537dup), leading to the existence of a severely truncated or absent protein due to nonsense-mediated mRNA decay (NMD) [28]. BRCA2 c.3865_3868del, c.5057T>A, and c.537dup variants were absent from the gnomAD (v2.1.1) and ALFA databases, while BRCA2 c.4284dup had a frequency of 1 out of 244426 alleles in the total population of gnomAD (v2.1.1) and was absent from the ALFA database (see Table 3). BRCA2 c.8243G>A had a frequency of 2/249060 in the total population of gnomAD (v2.1.1) and 1/25340 in ALFA. Various functional studies show a loss of function and/or protein stability linked to the BRCA2 c.8243G>A variant [29,30]. All of the BRCA2 variants were previously described as pathogenic [22,31,32,33].
Table 3.
Characteristics of the variants identified in the patients.
The missense ATM c.8494C>T variant was located in exon 58 out of 63, was present in 7 out 236730 alleles in the total population in gnomAD (v2.1.1), and has been previously described as pathogenic and associated with an increased cancer risk [34]. The ATM c.7516-2A>G variant located in intron 50 out of 62 has not been previously reported, and was not present in the gnomAD (v2.1.1) or ALFA databases. However, the variant was located in a region of the gene where other variants have been described as pathogenic, affecting a conserved splice site [35]. ATM c.7516-2A>G in silico evaluation results showed splicing alteration by wild-type acceptor site breakage. The nonsense ATM c.6326G>A variant in exon 43 out of 63 was predicted to cause loss-of-function by premature protein truncation or NMD. This variant was not found in the gnomAD (v2.1.1) or ALFA databases and has been previously reported as pathogenic [36].
Nonsense BRCA1 c.1121del variant caused a frameshift with a predicted stop codon two amino acids after the deletion, which could result in loss of normal protein function through protein truncation or NMD. This variant was absent in gnomAD (v2.1.1) or ALFA, but was present in several individuals suffering from breast and/or ovarian cancer [37]. This variant was also known as c.1240delC in the literature.
Missense CHEK2 c.499G>A variant leads to a substitution of a highly conserved amino acid. This variant was present in the total population of gnomAD (v2.1.1) in 6 out of 251424 alleles, and 3/100662 alleles in ALFA. Additionally, functional analysis showed a loss of function of the protein due to structural instability [38] or phosphorylation anomaly [39]. The in silico analysis of the variant predicted a deleterious effect on the protein, and CHEK2 loss-of-function variants are known to be pathogenic [40].
4. Discussion
PVs in BRCA1, BRCA2, CHEK2, and ATM increase the lifetime cancer risk of breast cancer [41]. In women carrying BRCA1 and BRCA2 PVs, the cumulative risk of breast cancer was 4% before the age of 30 for each gene, and reached 72% for BRCA1 and 69% for BRCA2 by age 80 [42]. For ATM variants, there was an estimated breast cancer relative risk of 2.8, and the absolute breast cancer risk reached 27% by 80 years. The CHEK2 breast cancer risk was variable for different PVs. Common CHEK2 truncating variants conferred a greater than twofold relative risk, while a less common I157T variant was associated with a 1.4-fold risk [43]. Similarly, in a study that included 65,057 women with breast cancer, the age of diagnosis of CHEK2 PV’s carriers was 47.7 years [41]. However, there is a lack of epidemiological data on BC risk in patients carrying PVs in two of these genes. Our study indicates very precocious and even metastatic BC in women with PVs in BRCA2 and ATM (patients 2 and 3) or BRCA2 and CHEK2 genes (patient 6), while a previous study evaluating 17 double-heterozygous patients with breast cancers failed to demonstrate a younger age at presentation in this group [13]. A similar trend could be expected when PVs in BRCA1 are associated with PVs in other CPGs.
The risks of other cancers are also elevated in BRCA1-, BRCA2-, and ATM-variant carriers. BRCA1 and BRCA2 PVs confer increased risks of prostate, pancreatic, and ovarian cancers [44], while moderate-to-high risks of pancreatic (OR 4.21), prostate (OR 2.58), and gastric (OR 2.97) cancers were estimated for ATM-variant carriers [24]. In our observations, two male patients were treated for a prostate cancer diagnosed at an early age, which might suggest that the BRCA2-linked risk is further increased by the presence of the ATM PV.
The reported pancreatic cancer risks in BRCA1 and BRCA2 carriers by the age of 70 years were 1.16% and 4.1% in men [44]. As BRCA1, BRCA2, and ATM proteins interact in the HR pathway, an additive effect on HR deficiency could be expected, giving a further increased risk of pancreatic cancer, as observed in patients 1, 4, and 5. Indeed, in a recent case report of a female patient carrying two heterozygous pathogenic variants in BRCA2 and ATM, breast cancer was diagnosed at 34 and pancreatic cancer at 48 years [45]. This raises the question of whether the previously-described reported young women with breast cancer (family 1) will need additional monitoring for their pancreatic cancer risk.
Therefore, our observations suggest that patients carrying a PV in BRCA2 plus another HR gene should be carefully monitored for BC, pancreatic cancer, and prostate cancer. However, incomplete penetrance and variability of the age of onset of the disease are also observed in double-heterozygous patients. In the second reported family, the proband’s father also carried both BRCA2 and ATM PVs (see Figure 1) but did not have any history of cancer, indicating that both genetic and non-genetic factors can influence cancer risk in variant carriers [44], while in the third family, the proband’s sister developed cancer at an older age, supporting the variable expressivity of these mutations. Further studies and larger cohorts are thus of course needed to better define the cancer risk associated with having two PVs in HR genes.
PVs in BRCA1 and BRCA2 have frequencies of 0.21% and 0.31% in the European population [46], while the frequencies of ATM and CHEK2 PVs reach 1% [47] and 1.4% [48]. These estimations, taken together, and given the scarcity of double-heterozygotes reports, indicate that the prevalence of digenic coinheritance is likely underestimated. Recently, even a patient with breast cancer and concurrent PVs in three cancer-related genes (BRCA1, BRCA2, and CHEK2) has been reported [49]. Therefore, given the high variability of phenotypes within families and between different families, when a cascade testing is performed after the identification of a familial PV, the assessment should not stop at the single known familial PV, at least in individuals with precocious breast, pancreatic, or prostate cancers; in those with multiple cancers; and in cases of cancers that are not frequently associated with the identified PV, as the possibility of co-segregation of another PV should not be neglected.
The size of the genetic panels used for cancer patients’ evaluation has progressively increased in recent years [50]. Consequently, the findings derived from these expanded panels are still in the preliminary stages, and it is impossible to directly compare the new data with previous results from shorter panels. Nonetheless, instances of double mutations are expected to remain relatively rare. After introducing multi-gene panel testing in 2014, by 2023, in the Fox Chase Cancer Center Risk Assessment Program Registry, 70 patients were found to carry at least two PVs in CPGs (excluding biallelic MUTYH PVs) [51]. In a review of 55,803 patients screened with a 25-gene hereditary cancer panel, 106 individuals (0.19%) showed PVs or likely pathogenic variants in two or more genes [52], a frequency of double heterozygotes very similar to that observed in the present study.
With the increase in patient numbers and the utilization of larger cohorts for analysis, more robust data will be available soon. Furthermore, the criteria for recommending genetic studies have undergone multiple revisions over time. Only recently has genetic testing for pancreatic and prostate cancer been included as part of the standard practice [53]. Consequently, the reports of larger cohorts of patients with diverse primary tumors will increase the likelihood of identifying cases with double mutations.
The small number of patients, the bias in recruitment, and the inability to evaluate the segregation in all of the families are the main limitations of our study. Additionally, we did not address the associated treatment strategies—platinum-based chemotherapy or PARP inhibitors—and the patients’ responses. With only six patients, we lack the data for meaningful comparisons or response-rate calculations. A larger study involving double-heterozygous patients is necessary to address these questions effectively.
Therefore, in young cancer patients from a family with a single known CPG PV, it could be useful to evaluate other genes to identify the potential transmission of several PVs and double-heterozygous carriers with a specific high cancer risk. Moreover, our data suggest that the surveillance of patients carrying two PVs in HR genes should include at least breast, pancreas, and prostate cancer screening, starting early. From our limited study, we would recommend starting a screening in those patients, at the latest, from the ages of 25, 40, and 50 for breast, prostate and pancreas cancer, respectively.
5. Conclusions
In conclusion, the early age of diagnosis and the development of multiple cancers in the reported patients indicate a very high risk of cancer in double-heterozygous patients associated with PVs in HR-related CPGs. Therefore, when a CPG PV is identified in a family, the usual cascade testing needs also to consider a study of other CPGs in patients with specific phenotypes, even distinct from other family members, either based on the age at diagnosis or the type of cancer.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jpm14060584/s1, Figure S1: Chromatograms of the variants identified in the patients; Table S1: DNA extraction; Table S2: Primer sequences used.
Author Contributions
Conceptualization, C.J. and V.B.; Data curation, M.V.F., M.M. and V.B.; Formal analysis, M.V.F., K.S., E.S., J.C. (Jérôme Coupier) and N.L.; Funding acquisition, V.B.; Investigation, M.V.F., M.M., H.R.K., P.W., J.C. (Joëlle Collignon), M.P. and O.P.; Methodology, M.V.F., M.M. and C.J.; Project administration, C.J. and V.B.; Resources, V.B.; Supervision, V.B.; Validation, V.B.; Writing—original draft, M.V.F. and V.B.; Writing—review and editing, M.V.F., M.M., K.S., E.S., N.L., H.R.K., P.W., J.C. (Joëlle Collignon), J.C. (Jérôme Coupier), M.P., O.P. and C.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Televie fellowship, grant number 7451419F; CHU Liège, grant number 981481200; and WALGEMED, grant number 1710180.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Comité d’Ethique Hospitalo-facultaire Universitaire de Liège (protocol code 2019/245 and date of approval 28 October 2019).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All the data relevant to the study is available in the manuscript.
Acknowledgments
CHU of Liege is member of the GENTURIS (GENetic TUmor RISk) European Reference Network.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Espenschied, C.R.; LaDuca, H.; Li, S.; McFarland, R.; Gau, C.-L.; Hampel, H. Multigene Panel Testing Provides a New Perspective on Lynch Syndrome. J. Clin. Oncol. 2017, 35, 2568–2575. [Google Scholar] [CrossRef] [PubMed]
- Esplin, E.D.; Nielsen, S.M.; Bristow, S.L.; Garber, J.E.; Hampel, H.; Rana, H.Q.; Samadder, N.J.; Shore, N.D.; Nussbaum, R.L. Universal Germline Genetic Testing for Hereditary Cancer Syndromes in Patients With Solid Tumor Cancer. JCO Precis. Oncol. 2022, 6, e2100516. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Couch, F.J. Germline Genetic Testing for Breast Cancer Risk: The Past, Present, and Future. Am. Soc. Clin. Oncol. Educ. Book. 2019, 39, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N. Mainstreaming genetic testing of cancer predisposition genes. Clin. Med. 2014, 14, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, K.D.; Obeid, E.; Daly, M.B.; Hall, M.J. Cascade Genetic Testing for Hereditary Cancer Risk: An Underutilized Tool for Cancer Prevention. JCO Precis. Oncol. 2021, 5, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Ashok, A.; Stoll, J.; Mauer, E.; Nepomuceno, V.M.; Blackwell, K.L.; Garber, J.E.; Meric-Bernstam, F. Prevalence of Germline Findings Among Tumors From Cancer Types Lacking Hereditary Testing Guidelines. JAMA Netw. Open 2022, 5, e2213070. [Google Scholar] [CrossRef]
- Kotsopoulos, J.; Hathaway, C.A.; Narod, S.A.; Teras, L.R.; Patel, A.V.; Hu, C.; Yadav, S.; Couch, F.J.; Tworoger, S.S. Germline Mutations in 12 Genes and Risk of Ovarian Cancer in Three Population-Based Cohorts. Cancer Epidemiol. Biomark. Prev. 2023, 32, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Caliendo, G.; D’Elia, G.; Resse, M.; Casamassimi, A.; Minucci, P.B.; Dello Ioio, C.; Cioffi, M.; Molinari, A.M. Five Italian Families with Two Mutations in BRCA Genes. Genes 2020, 11, 1451. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Bian, J.; Qian, X.; Shao, L.; Li, H.; Zhang, L.; Wang, L. Case Report: Coinheritance of Germline Mutations in APC and BRCA1 in Colorectal Cancer. Front. Oncol. 2021, 11, 658389. [Google Scholar] [CrossRef]
- Andrés, R.; Menao, S.; Arruebo, M.; Quílez, E.; Cardiel, M.J. Double heterozygous mutation in the BRCA1 and ATM genes involved in development of primary metachronous tumours: A case report. Breast Cancer Res. Treat. 2019, 177, 767–770. [Google Scholar] [CrossRef]
- Slaught, C.; Berry, E.G.; Bacik, L.; Skalet, A.H.; Anadiotis, G.; Tuohy, T.; Leachman, S.A. Clinical challenges in interpreting multiple pathogenic mutations in single patients. Hered. Cancer Clin. Pract. 2021, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Lavie, O.; Narod, S.; Lejbkowicz, F.; Dishon, S.; Goldberg, Y.; Gemer, O.; Rennert, G. Double heterozygosity in the BRCA1 and BRCA2 genes in the Jewish population. Ann. Oncol. 2011, 22, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Sokolenko, A.P.; Bogdanova, N.; Kluzniak, W.; Preobrazhenskaya, E.V.; Kuligina, E.S.; Iyevleva, A.G.; Aleksakhina, S.N.; Mitiushkina, N.V.; Gorodnova, T.V.; Bessonov, A.A.; et al. Double heterozygotes among breast cancer patients analyzed for BRCA1, CHEK2, ATM, NBN/NBS1, and BLM germ-line mutations. Breast Cancer Res. Treat. 2014, 145, 553–562. [Google Scholar] [CrossRef]
- Helleday, T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis 2010, 31, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, L.; Piombino, C.; Toss, A. Germline Mutations in Other Homologous Recombination Repair-Related Genes Than BRCA1/2: Predictive or Prognostic Factors? J. Pers. Med. 2021, 11, 245. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.K. Cancer Risk and the ATM Gene: A Continuing Debate. JNCI J. Natl. Cancer Inst. 2000, 92, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Hirasawa, A. Homologous Recombination Deficiencies and Hereditary Tumors. Int. J. Mol. Sci. 2021, 23, 348. [Google Scholar] [CrossRef] [PubMed]
- Jonathan, L.; Philipp, H.; Charlie, G.; Michael, F.; Ignace, V.; Gordon, R.; Clare, S.; Werner, M.; Ronnie, S.-F.; Tamar, S.; et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef]
- Moiseyenko, V.M.; Chubenko, V.A.; Moiseyenko, F.V.; Zhabina, A.S.; Gorodnova, T.V.; Komarov, Y.I.; Bogdanov, A.A.; Sokolenko, A.P.; Imyanitov, E.N. Evidence for clinical efficacy of mitomycin C in heavily pretreated ovarian cancer patients carrying germ-line BRCA1 mutation. Med. Oncol. 2014, 31, 199. [Google Scholar] [CrossRef]
- Conroy, M.; Borad, M.J.; Bryce, A.H. Hypoxia-Activated Alkylating Agents in BRCA1-Mutant Ovarian Serous Carcinoma. Cureus 2017, 9, e1517. [Google Scholar] [CrossRef] [PubMed]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation–Positive Women With Ovarian Cancer: A Report From the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Nazarali, S.; Narod, S.A. Multiple primary cancers as a guide to heritability. Int. J. Cancer 2014, 135, 1756–1763. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.J.; Bernhisel, R.; Hughes, E.; Larson, K.; Rosenthal, E.T.; Singh, N.A.; Lancaster, J.M.; Kurian, A.W. Germline pathogenic variants in the Ataxia Telangiectasia Mutated (ATM) gene are associated with high and moderate risks for multiple cancers. Cancer Prev. Res. 2021, 14, 433–440. [Google Scholar] [CrossRef]
- Harvengt, J.; Lumaka, A.; Fasquelle, C.; Caberg, J.H.; Mastouri, M.; Janssen, A.; Palmeira, L.; Bours, V. HIDEA syndrome: A new case report highlighting similarities with ROHHAD syndrome. Front. Genet. 2023, 14, 1137767. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Ware, M.D.; DeSilva, D.; Sinilnikova, O.M.; Stoppa-Lyonnet, D.; Tavtigian, S.V.; Mazoyer, S. Does nonsense-mediated mRNA decay explain the ovarian cancer cluster region of the BRCA2 gene? Oncogene 2006, 25, 323–328. [Google Scholar] [CrossRef]
- Richardson, M.E.; Hu, C.; Lee, K.Y.; LaDuca, H.; Fulk, K.; Durda, K.M.; Deckman, A.M.; Goldgar, D.E.; Monteiro, A.N.A.; Gnanaolivu, R.; et al. Strong functional data for pathogenicity or neutrality classify BRCA2 DNA-binding-domain variants of uncertain significance. Am. J. Hum. Genet. 2021, 108, 458–468. [Google Scholar] [CrossRef]
- Guidugli, L.; Pankratz, V.S.; Singh, N.; Thompson, J.; Erding, C.A.; Engel, C.; Schmutzler, R.; Domchek, S.; Nathanson, K.; Radice, P.; et al. A classification model for BRCA2 DNA binding domain missense variants based on homology directed repair activity. Cancer Res. 2013, 73, 265–275. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Hamann, U.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.-H.; et al. Mutational Spectrum in a Worldwide Study of 29,700 Families with BRCA1 or BRCA2 Mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Laitman, Y.; Friebel, T.M.; Yannoukakos, D.; Fostira, F.; Konstantopoulou, I.; Figlioli, G.; Bonanni, B.; Manoukian, S.; Zuradelli, M.; Tondini, C.; et al. The spectrum of BRCA1 and BRCA2 pathogenic sequence variants in Middle Eastern, North African, and South European countries. Hum. Mutat. 2019, 40, e1–e23. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Tozkir, H.; Gurkan, H.; Atli, E.I.; Yalcintepe, S.; Atli, E.; Sezer, A.; Eker, D.; Tuncbilek, N.; Tastekin, E.; et al. Genetic screening results of individuals with high risk BRCA- related breast/ovarian cancer in Trakya region of Turkey. J. BUON 2020, 25, 1337–1347. [Google Scholar] [PubMed]
- Mitui, M.; Nahas, S.; Du, L.; Yang, Z.; Lai, C.; Nakamura, K.; Arroyo, S.; Scott, S.; Purayidom, A.; Concannon, P.; et al. Functional and Computational Assessment of Missense Variants in the Ataxia-Telangiectasia Mutated (ATM) Gene: Mutations with Increased Cancer Risk. Hum. Mutat. 2009, 30, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Delia, D.; Mizutani, S.; Panigone, S.; Tagliabue, E.; Fontanella, E.; Asada, M.; Yamada, T.; Taya, Y.; Prudente, S.; Saviozzi, S.; et al. ATM protein and p53-serine 15 phosphorylation in ataxia-telangiectasia (AT) patients and at heterozygotes. Br. J. Cancer 2000, 82, 1938–1945. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, S.; Pozzi, E.; Gatti, R.A.; Brusco, A. Deep-intronic ATM mutation detected by genomic resequencing and corrected in vitro by antisense morpholino oligonucleotide (AMO). Eur. J. Hum. Genet. 2013, 21, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Borg, Å.; Haile, R.W.; Malone, K.E.; Capanu, M.; Diep, A.; Törngren, T.; Teraoka, S.; Begg, C.B.; Thomas, D.C.; Concannon, P.; et al. Characterization of BRCA1 and BRCA2 Deleterious Mutations and Variants of Unknown Clinical Significance in Unilateral and Bilateral Breast Cancer: The WECARE Study. Hum. Mutat. 2010, 31, E1200–E1240. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Williams, B.L.; Haire, L.F.; Goldberg, M.; Wilker, E.; Durocher, D.; Yaffe, M.B.; Jackson, S.P.; Smerdon, S.J. Structural and Functional Versatility of the FHA Domain in DNA-Damage Signaling by the Tumor Suppressor Kinase Chk2. Mol. Cell 2002, 9, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Boonen, R.A.C.M.; Wiegant, W.W.; Celosse, N.; Vroling, B.; Heijl, S.; Kote-Jarai, Z.; Mijuskovic, M.; Cristea, S.; Solleveld-Westerink, N.; van Wezel, T.; et al. Functional Analysis Identifies Damaging CHEK2 Missense Variants Associated with Increased Cancer Risk. Cancer Res. 2022, 82, 615–631. [Google Scholar] [CrossRef]
- Cybulski, C.; Wokołorczyk, D.; Jakubowska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Masojć, B.; Dębniak, T.; Górski, B.; Blecharz, P.; et al. Risk of Breast Cancer in Women With a CHEK2 Mutation With and Without a Family History of Breast Cancer. J. Clin. Oncol. 2011, 29, 3747–3752. [Google Scholar] [CrossRef]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Graffeo, R.; Rana, H.Q.; Conforti, F.; Bonanni, B.; Cardoso, M.J.; Paluch-Shimon, S.; Pagani, O.; Goldhirsch, A.; Partridge, A.H.; Lambertini, M.; et al. Moderate penetrance genes complicate genetic testing for breast cancer diagnosis: ATM, CHEK2, BARD1 and RAD51D. Breast 2022, 65, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Friedman, E. Cancer risks among BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2007, 96, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Duzkale Teker, N.; Eyerci, N. Double Heterozygous Mutations in the BRCA2 and ATM Genes: A Case Report and Review of the Literature. Breast Care 2021, 16, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Domchek, S.M.; Nathanson, K.L.; Robson, M.E. Population Frequency of Germline BRCA1/2 Mutations. J. Clin. Oncol. 2016, 34, 4183–4185. [Google Scholar] [CrossRef] [PubMed]
- Swift, M.; Morrell, D.; Cromartie, E.; Chamberlin, A.R.; Skolnick, M.H.; Bishop, D.T. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am. J. Hum. Genet. 1986, 39, 573–583. [Google Scholar]
- Nguyen-Dumont, T.; Dowty, J.G.; Steen, J.A.; Renault, A.-L.; Hammet, F.; Mahmoodi, M.; Theys, D.; Rewse, A.; Tsimiklis, H.; Winship, I.M.; et al. Population-Based Estimates of the Age-Specific Cumulative Risk of Breast Cancer for Pathogenic Variants in CHEK2: Findings from the Australian Breast Cancer Family Registry. Cancers 2021, 13, 1378. [Google Scholar] [CrossRef]
- Sukumar, J.; Kassem, M.; Agnese, D.; Pilarski, R.; Ramaswamy, B.; Sweet, K.; Sardesai, S. Concurrent germline BRCA1, BRCA2, and CHEK2 pathogenic variants in hereditary breast cancer: A case series. Breast Cancer Res. Treat. 2021, 186, 569–575. [Google Scholar] [CrossRef]
- Wang, A.; Everett, J.N.; Chun, J.; Cen, C.; Simeone, D.M.; Schnabel, F. Impact of changing guidelines on genetic testing and surveillance recommendations in a contemporary cohort of breast cancer survivors with family history of pancreatic cancer. Sci. Rep. 2021, 11, 12491. [Google Scholar] [CrossRef]
- Hall, M.J.; McSweeny, M.J.; Rainey, K.; Campbell, H.; Nguyen, C.; Neumann, C. Risks and implications of multiple actionable pathogenic germline variants discovered by panel-based cancer predisposition testing. J. Clin. Oncol. 2023, 41, 792. [Google Scholar] [CrossRef]
- Weitzel, J.N.; Blazer, K.R.; Nehoray, B.; Kidd, J.; Slavin, T.P.; Solomon, I.; Niell-Swiller, M.; Rybak, C.; Saam, J. Assessment of the clinical presentation of patients with at least two deleterious mutations on multi-gene panel testing. J. Clin. Oncol. 2015, 33, 1514. [Google Scholar] [CrossRef]
- Dal Buono, A.; Poliani, L.; Greco, L.; Bianchi, P.; Barile, M.; Giatti, V.; Bonifacio, C.; Carrara, S.; Malesci, A.; Laghi, L. Prevalence of Germline Mutations in Cancer Predisposition Genes in Patients with Pancreatic Cancer or Suspected Related Hereditary Syndromes: Historical Prospective Analysis. Cancers 2023, 15, 1852. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).