
Major Causes of Conflicting Interpretations of Variant Pathogenicity in Rare Disease: A Systematic Analysis



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genetic Variant Data
2.2. Analysis of Genes for Enrichment of Genetic Variants with COIs
2.3. Analysis of Functional Properties of Genes Bearing COI Variants
2.4. Statistical Analysis
3. Results
3.1. Common Properties of Variants with Conflicting Interpretations of Pathogenicity
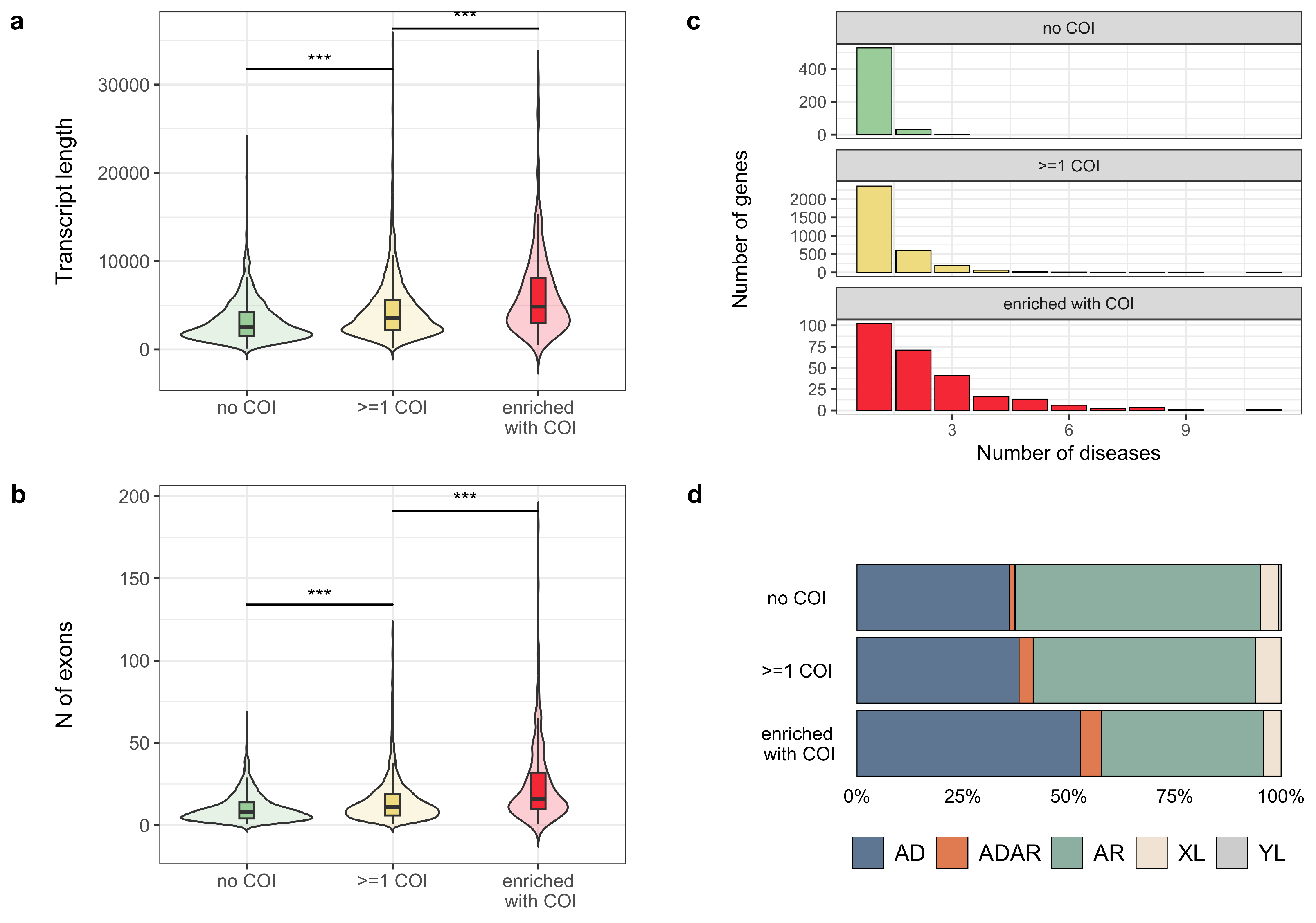
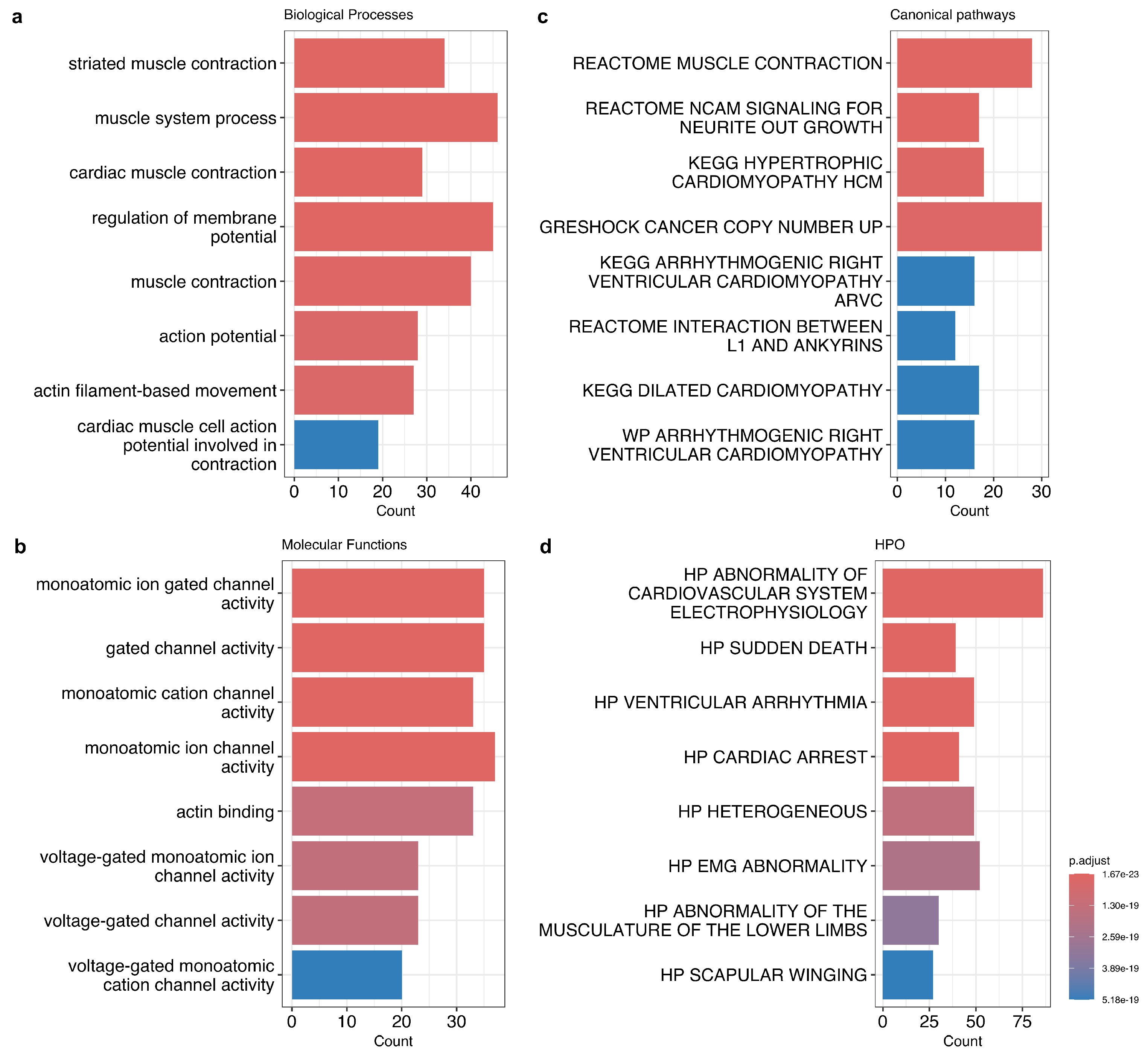
3.2. Properties of Genes with a High Proportion of COI Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| COI | conflicting interpretations of pathogenicity |
| HPO | Human Phenotype Ontology |
| NGS | next-generation sequencing |
| OMIM | Online Mendelian Inheritance in Man |
References
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Barbitoff, Y.A.; Ushakov, M.O.; Lazareva, T.E.; Nasykhova, Y.A.; Glotov, A.S.; Predeus, A.V. Bioinformatics of germline variant discovery for rare disease diagnostics: Current approaches and remaining challenges. Briefings Bioinform. 2024, 25, bbad508. [Google Scholar] [CrossRef]
- Federici, G.; Soddu, S. Variants of uncertain significance in the era of high-throughput genome sequencing: A lesson from breast and ovary cancers. J. Exp. Clin. Cancer Res. 2020, 39, 46. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.M.; Dolinsky, J.S.; Knight Johnson, A.E.; Pesaran, T.; Azzariti, D.R.; Bale, S.; Chao, E.C.; Das, S.; Vincent, L.; Rehm, H.L. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet. Med. 2017, 19, 1096–1104. [Google Scholar] [CrossRef]
- Bland, A.; Harrington, E.A.; Dunn, K.; Pariani, M.; Platt, J.C.; Grove, M.E.; Caleshu, C. Clinically impactful differences in variant interpretation between clinicians and testing laboratories: A single-center experience. Genet. Med. 2018, 20, 369–373. [Google Scholar] [CrossRef]
- Smirnova, O.; Lagutinskaya, D. The role of polymorphisms of PNPLA3, MBOAT7, and TM6SF2 in the development of non-alcoholic fatty liver disease in metabolic syndrome. Obes. Metab. 2022, 19, 166–170. [Google Scholar] [CrossRef]
- Stepanova, A.; Abrukova, A.; Savaskina, E.; Polyakov, A. Mutation p. E92K is the primary cause of cystic fibrosis in Chuvashes. Russ. J. Genet. 2012, 48, 731–737. [Google Scholar] [CrossRef]
- Petrova, N.; Balinova, N.; Marakhonov, A.; Vasilyeva, T.; Kashirskaya, N.; Galkina, V.; Ginter, E.; Kutsev, S.; Zinchenko, R. Ethnic differences in the frequency of CFTR gene mutations in populations of the European and North Caucasian Part of the Russian Federation. Front. Genet. 2021, 12, 678374. [Google Scholar] [CrossRef]
- Ideozu, J.E.; Liu, M.; Riley-Gillis, B.M.; Paladugu, S.R.; Rahimov, F.; Krishnan, P.; Tripathi, R.; Dorr, P.; Levy, H.; Singh, A.; et al. Diversity of CFTR variants across ancestries characterized using 454,727 UK biobank whole exome sequences. Genome Med. 2024, 16, 43. [Google Scholar] [CrossRef]
- Tian, X.; Liu, Y.; Yang, J.; Wang, H.; Liu, T.; Xu, W.; Li, X.; Zhu, Y.; Xu, K.F.; Zhang, X. p. G970D is the most frequent CFTR mutation in Chinese patients with cystic fibrosis. Hum. Genome Var. 2016, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.A.; Polev, D.E.; Glotov, A.S.; Serebryakova, E.A.; Shcherbakova, I.V.; Kiselev, A.M.; Kostareva, A.A.; Glotov, O.S.; Predeus, A.V. Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Sci. Rep. 2020, 10, 2057. [Google Scholar] [CrossRef]
- Belova, V.; Vasiliadis, I.; Repinskaia, Z.; Samitova, A.; Shmitko, A.; Ponikarovskaya, N.; Suchalko, O.; Cheranev, V.; Shatalov, P.; Shegai, P.; et al. Comparative evaluation of four exome enrichment solutions in 2024: Agilent, Roche, Vazyme and Nanodigmbio. bioRxiv 2024. [Google Scholar] [CrossRef]
- Barbitoff, Y.A.; Abasov, R.; Tvorogova, V.E.; Glotov, A.S.; Predeus, A.V. Systematic benchmark of state-of-the-art variant calling pipelines identifies major factors affecting accuracy of coding sequence variant discovery. BMC Genom. 2022, 23, 155. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, A.; Vélez-Santamaría, V.; Verdura, E.; Rodríguez-Palmero, A.; Ruiz, M.; Fourcade, S.; Planas-Serra, L.; Launay, N.; Guilera, C.; Martínez, J.J.; et al. ClinPrior: An algorithm for diagnosis and novel gene discovery by network-based prioritization. Genome Med. 2023, 15, 68. [Google Scholar] [CrossRef]
- Jacobsen, J.O.; Kelly, C.; Cipriani, V.; Research Consortium, G.E.; Mungall, C.J.; Reese, J.; Danis, D.; Robinson, P.N.; Smedley, D. Phenotype-driven approaches to enhance variant prioritization and diagnosis of rare disease. Hum. Mutat. 2022, 43, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, J.P.; Bartos, D.C.; Kapplinger, J.D.; Tester, D.J.; Delisle, B.P.; Ackerman, M.J. The promise and peril of precision medicine: Phenotyping still matters most. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2016; Volume 91, pp. 1606–1616. [Google Scholar]
- Gaba, P.; Bos, J.M.; Cannon, B.C.; Cha, Y.M.; Friedman, P.A.; Asirvatham, S.J.; Ackerman, M.J. Implantable cardioverter-defibrillator explantation for overdiagnosed or overtreated congenital long QT syndrome. Heart Rhythm 2016, 13, 879–885. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The human phenotype ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.N.; Köhler, S.; Bauer, S.; Seelow, D.; Horn, D.; Mundlos, S. The Human Phenotype Ontology: A tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008, 83, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Lincoln, S.E.; Kobayashi, Y.; Nykamp, K.; Nussbaum, R.L.; Topper, S. Sources of discordance among germ-line variant classifications in ClinVar. Genet. Med. 2017, 19, 1118–1126. [Google Scholar] [CrossRef]
- Makhnoon, S.; Levin, B.; Ensinger, M.; Mattie, K.; Volk, R.J.; Zhao, Z.; Mendoza, T.; Shete, S.; Samiian, L.; Grana, G.; et al. A multicenter study of clinical impact of variant of uncertain significance reclassification in breast, ovarian and colorectal cancer susceptibility genes. Cancer Med. 2023, 12, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Weck, K.E. Interpretation of genomic sequencing: Variants should be considered uncertain until proven guilty. Genet. Med. 2018, 20, 291–293. [Google Scholar] [CrossRef]
- Morales, J.; Pujar, S.; Loveland, J.E.; Astashyn, A.; Bennett, R.; Berry, A.; Cox, E.; Davidson, C.; Ermolaeva, O.; Farrell, C.M.; et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 2022, 604, 310–315. [Google Scholar] [CrossRef]
- Rosamilia, M.B.; Markunas, A.M.; Kishnani, P.S.; Landstrom, A.P. Underrepresentation of Diverse Ancestries Drives Uncertainty in Genetic Variants Found in Cardiomyopathy-Associated Genes. JACC Adv. 2024, 3, 100767. [Google Scholar] [CrossRef]
- Cerrone, M.; Remme, C.A.; Tadros, R.; Bezzina, C.R.; Delmar, M. Beyond the one gene–one disease paradigm: Complex genetics and pleiotropy in inheritable cardiac disorders. Circulation 2019, 140, 595–610. [Google Scholar] [CrossRef]
- Mestroni, L. Phenotypic heterogeneity of sarcomeric gene mutations: A matter of gain and loss? J. Am. Coll. Cardiol. 2009, 54, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Remme, C.A. SCN5A channelopathy: Arrhythmia, cardiomyopathy, epilepsy and beyond. Philos. Trans. R. Soc. 2023, 378, 20220164. [Google Scholar] [CrossRef] [PubMed]
- Ciconte, G.; Monasky, M.M.; Santinelli, V.; Micaglio, E.; Vicedomini, G.; Anastasia, L.; Negro, G.; Borrelli, V.; Giannelli, L.; Santini, F.; et al. Brugada syndrome genetics is associated with phenotype severity. Eur. Heart J. 2020, 42, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Lazareva, T.E.; Barbitoff, Y.A.; Nasykhova, Y.A.; Pavlova, N.S.; Bogaychuk, P.M.; Glotov, A.S. Statistical Dissection of the Genetic Determinants of Phenotypic Heterogeneity in Genes with Multiple Associated Rare Diseases. Genes 2023, 14, 2100. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazareva, T.E.; Barbitoff, Y.A.; Nasykhova, Y.A.; Glotov, A.S. Major Causes of Conflicting Interpretations of Variant Pathogenicity in Rare Disease: A Systematic Analysis. J. Pers. Med. 2024, 14, 864. https://doi.org/10.3390/jpm14080864
Lazareva TE, Barbitoff YA, Nasykhova YA, Glotov AS. Major Causes of Conflicting Interpretations of Variant Pathogenicity in Rare Disease: A Systematic Analysis. Journal of Personalized Medicine. 2024; 14(8):864. https://doi.org/10.3390/jpm14080864
Chicago/Turabian StyleLazareva, Tatyana E., Yury A. Barbitoff, Yulia A. Nasykhova, and Andrey S. Glotov. 2024. "Major Causes of Conflicting Interpretations of Variant Pathogenicity in Rare Disease: A Systematic Analysis" Journal of Personalized Medicine 14, no. 8: 864. https://doi.org/10.3390/jpm14080864
APA StyleLazareva, T. E., Barbitoff, Y. A., Nasykhova, Y. A., & Glotov, A. S. (2024). Major Causes of Conflicting Interpretations of Variant Pathogenicity in Rare Disease: A Systematic Analysis. Journal of Personalized Medicine, 14(8), 864. https://doi.org/10.3390/jpm14080864