
Innovative Strategies in X-ray Crystallography for Exploring Structural Dynamics and Reaction Mechanisms in Metabolic Disorders
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
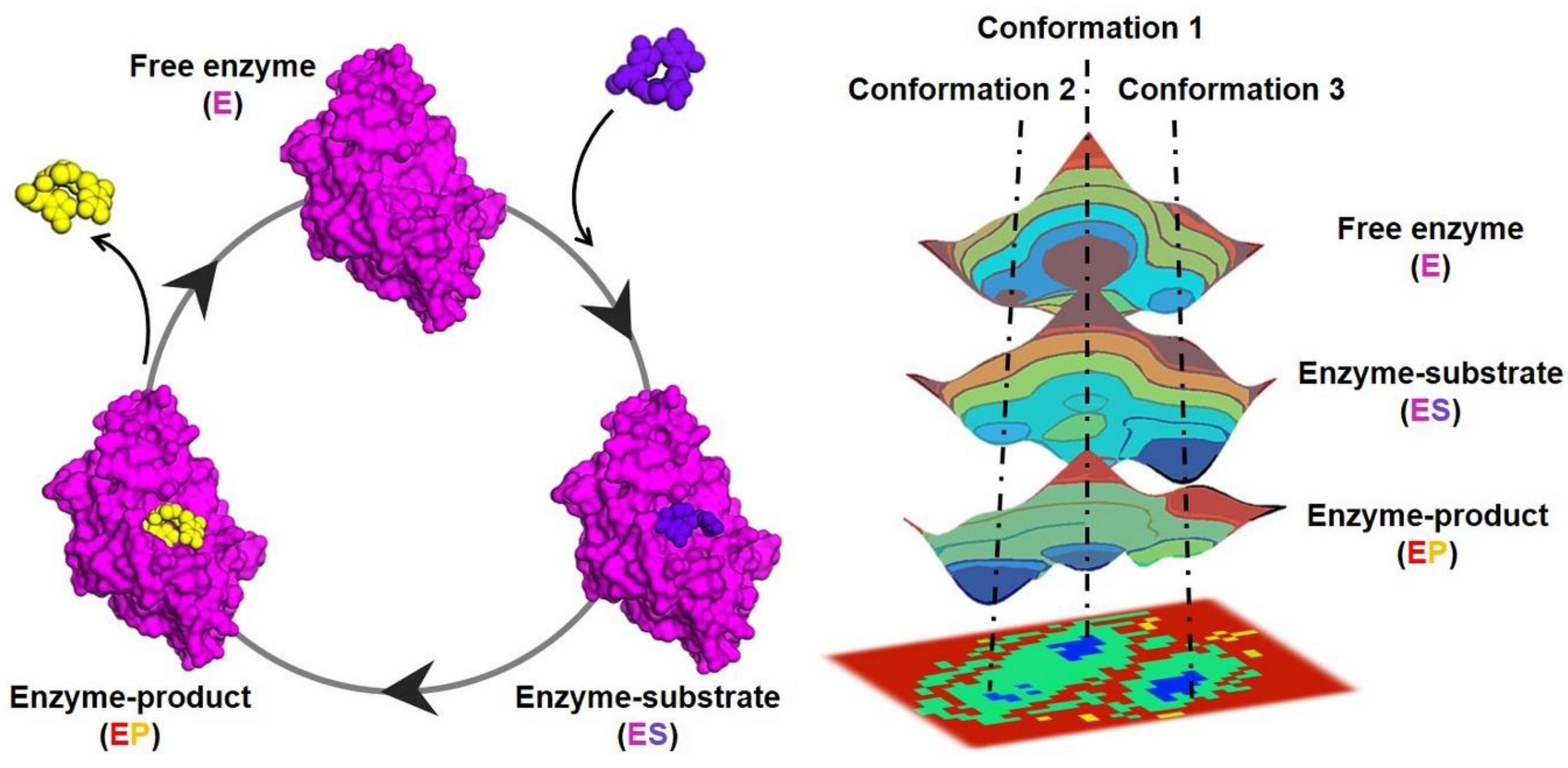
:1. The Role of Enzyme Dynamics in Metabolic Diseases
2. Structural Biology Approaches to the Study of Enzyme Dynamics in Metabolic Diseases
3. X-ray Crystallography and Its Role in Enzyme Dynamics
4. Microcrystals Are Ideal for Studying Protein Dynamics and Catalysis of Metabolic Diseases
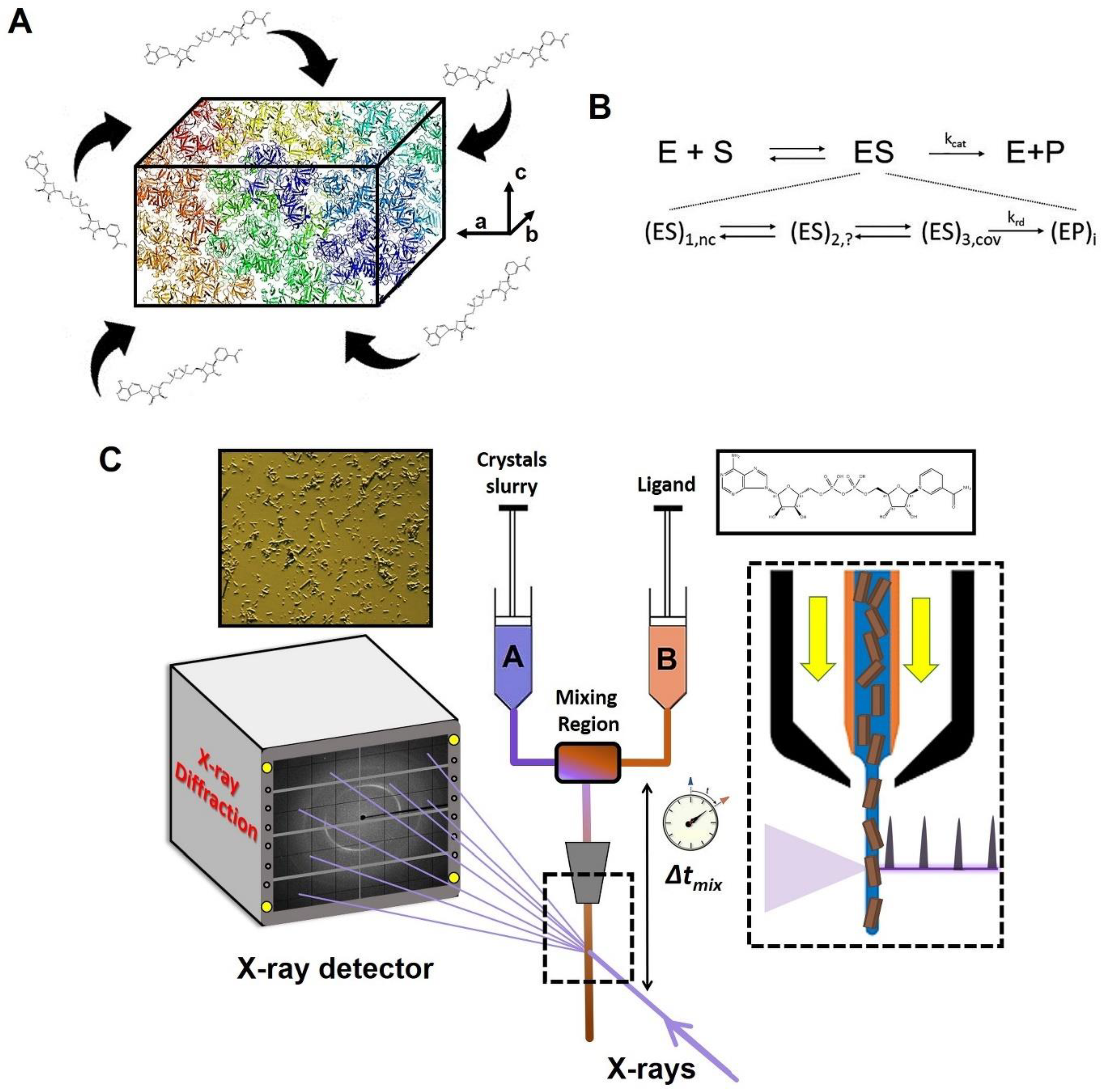
5. Time-Resolved Serial Crystallography
6. Metabolic Reactions with Time-Resolved Serial Crystallography
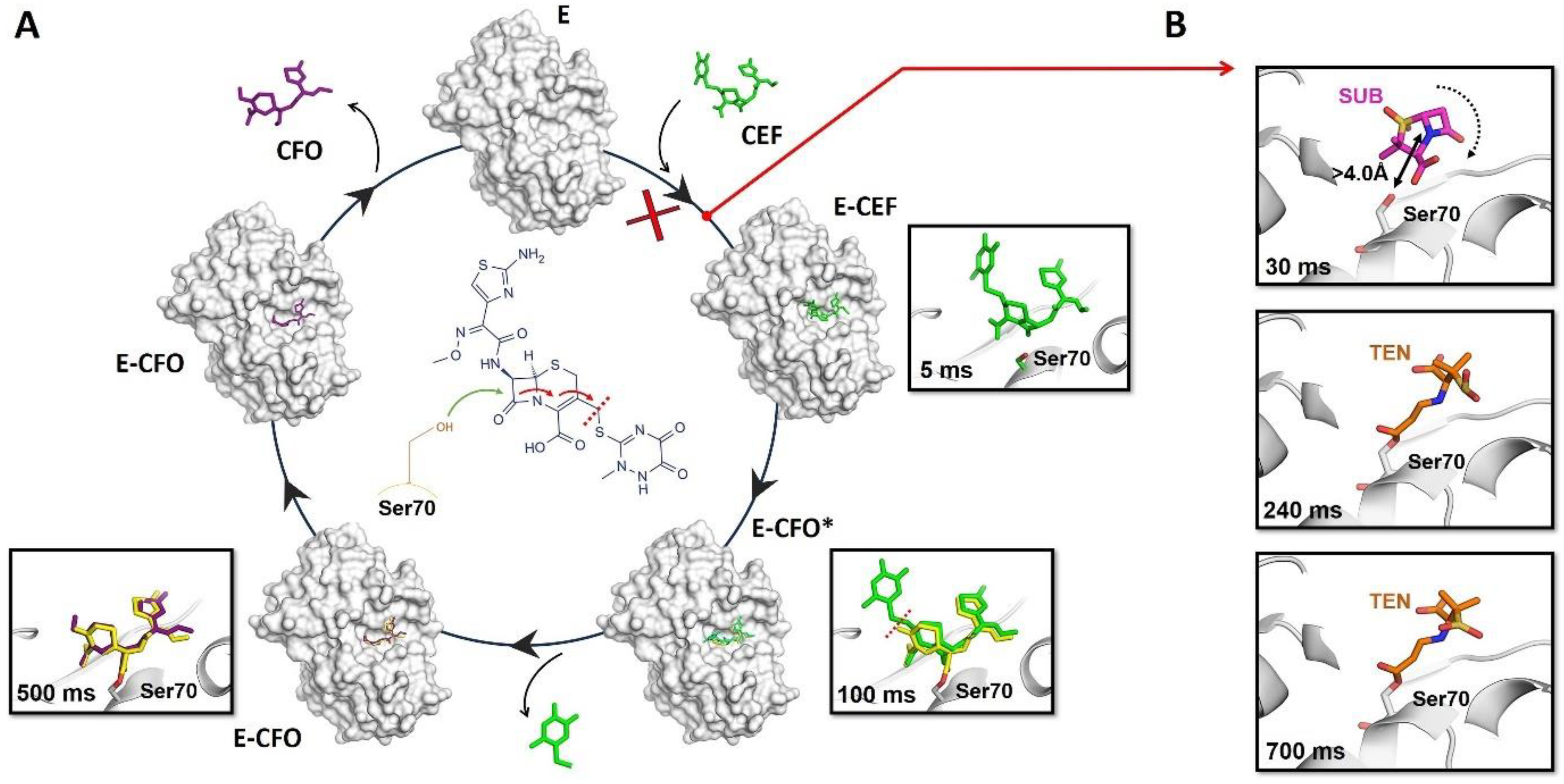
7. Examples of How Time-Resolved Serial Crystallography Studies Can Help Fight Metabolic Diseases
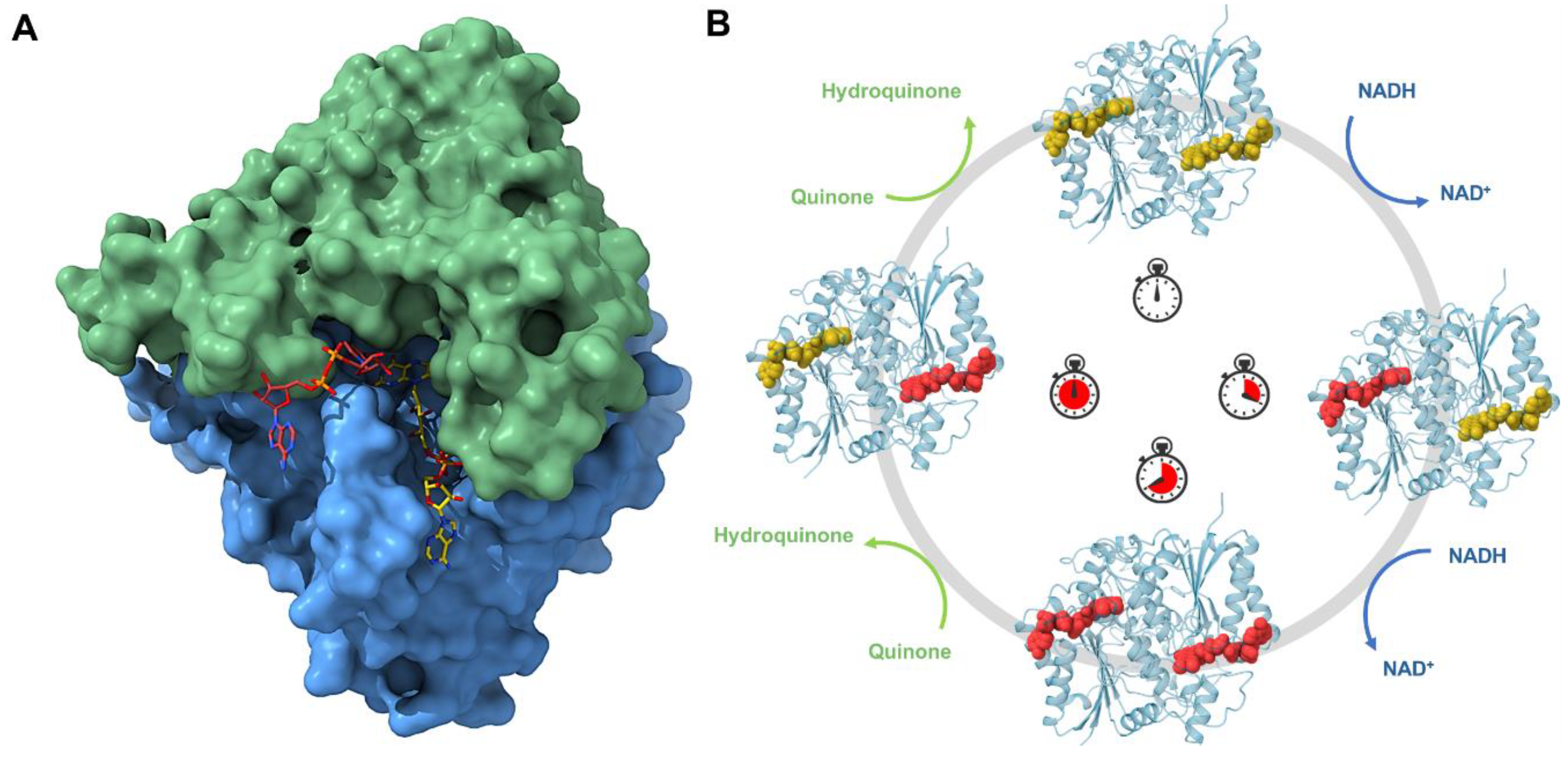
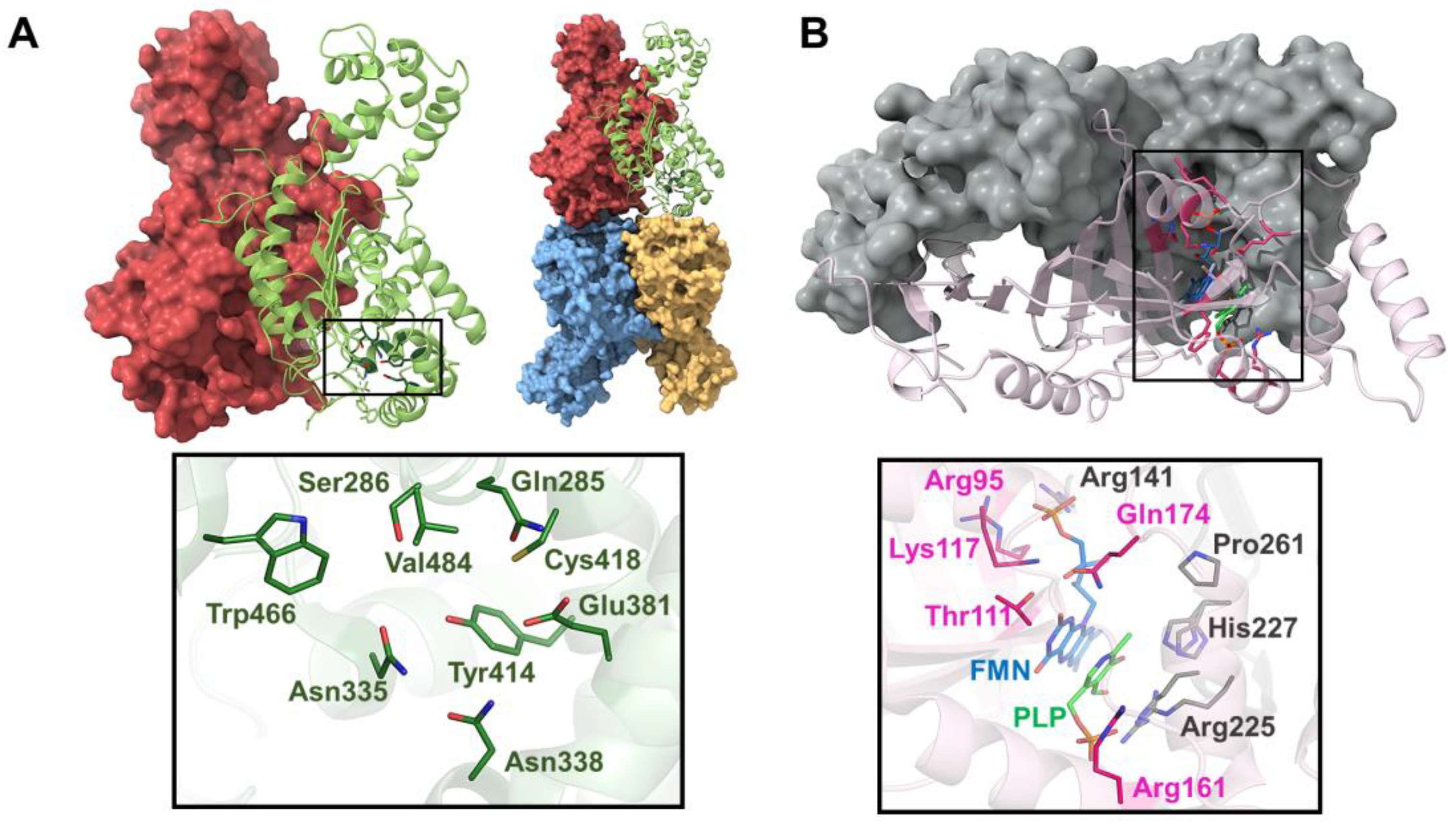
7.1. Human NAD(P)H:Quinone Oxidoreductase 1 (NQO1)
7.2. Glutaminase C (GAC)
7.3. G Protein-Coupled Receptors (GPCRs)
7.4. Metalloenzymes
7.4.1. Metalloenzymes and Crystallography
7.4.2. Metalloenzymes in Metabolic Diseases
- Capturing Transient States: Metalloenzymes often undergo conformational changes and structural rearrangements during catalytic cycles or upon substrate binding. Capturing the transient states and intermediate structures of metalloenzymes with high temporal resolution will provide us with insights into reaction mechanisms and dynamics.
- Visualizing Metal Ion Coordination: Metal ions are crucial to the catalytic mechanisms of metalloenzymes, influencing coordination geometry, ligand-binding dynamics, and redox states. Understanding these factors is essential for gaining insights into the enzymes’ catalytic activities and regulatory mechanisms.
- Probing Metallocofactor Dynamics: Many metalloenzymes feature metal cofactors such as iron-sulfur clusters, heme groups, or metal ions that undergo redox reactions or structural changes during enzyme turnover. Tracking the dynamics of these metallocofactors in real time can reveal how they interact with substrates, cofactors, and inhibitors during catalysis.
- Uncovering Allosteric Regulation: Metalloenzymes are often regulated by allosteric effectors or environmental factors that influence their activity and specificity. Understanding how allosteric communication pathways and conformational changes are triggered by ligand binding or environmental cues is essential for gaining insights into enzyme regulation and function.
- Drug Discovery and Design: Metalloenzymes are prime targets for drug discovery due to their critical roles in various biological processes and disease pathways. Revealing atomic-level details of enzyme-substrate interactions, inhibitor-binding modes, and allosteric sites can significantly aid in structure-based drug design, guiding the development of selective and potent enzymatic inhibitors for therapeutic intervention.
- Understanding Disease Mechanisms: Dysregulations or mutations in metalloenzymes are linked to various diseases, including cancer, neurodegenerative disorders, and metabolic conditions. Understanding the structural and dynamic consequences of these disease-associated mutations can offer mechanistic insights into disease pathology and identify potential targets for therapeutic intervention.
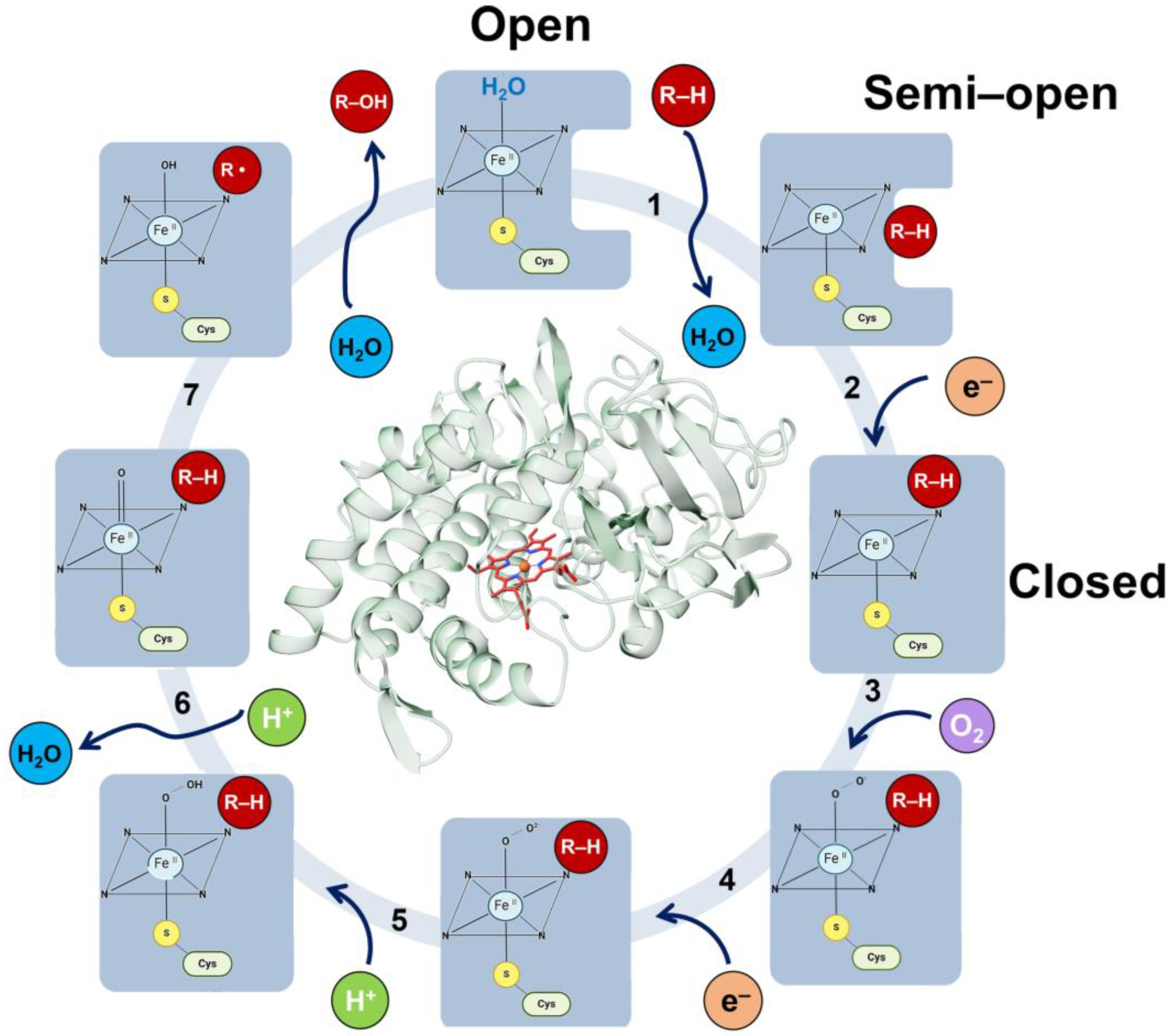
7.4.3. The Human Metalloenzyme Cytochrome P450 2D6 (CYP2D6)
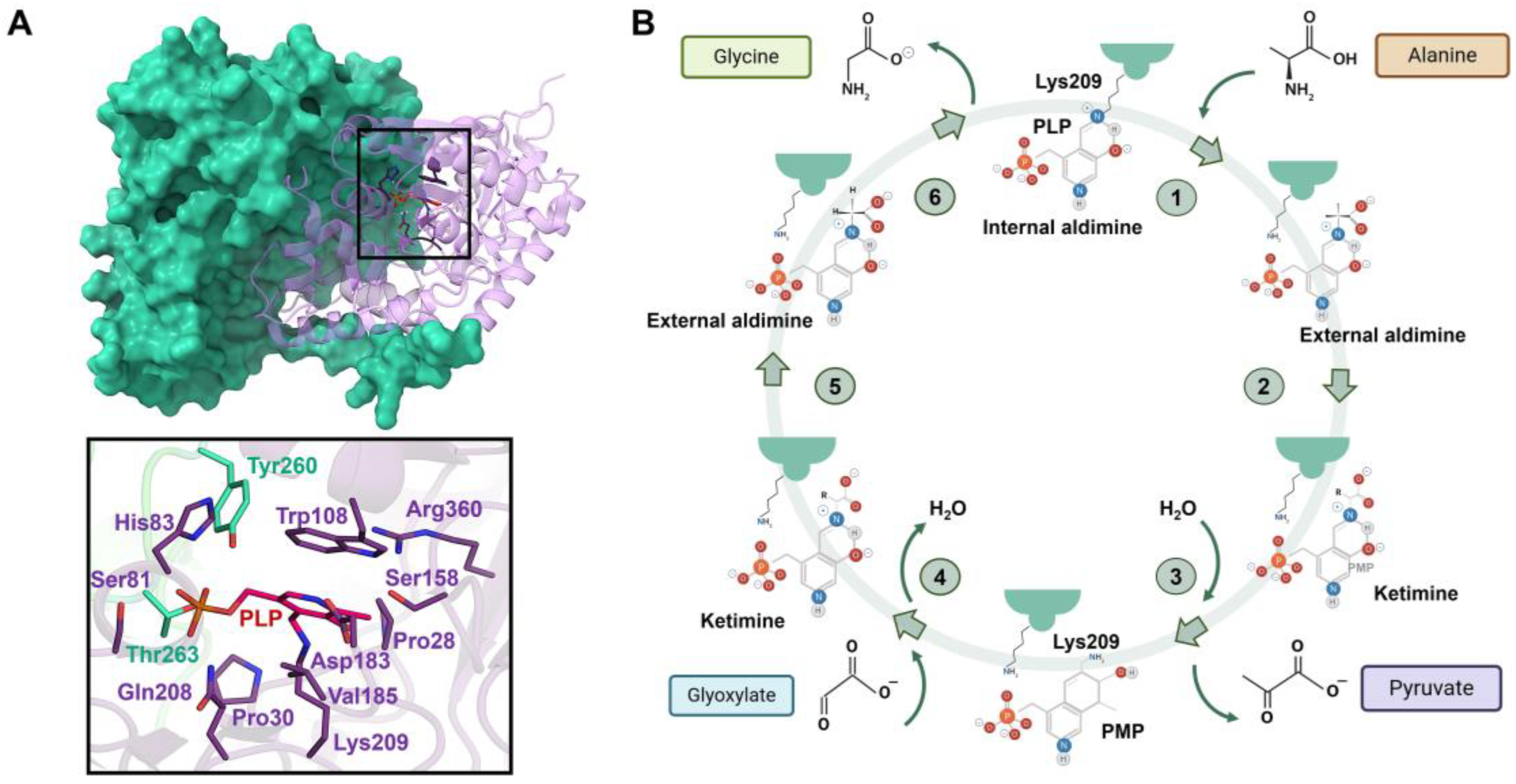
7.5. Human Alanine-Glyoxylate Aminotransferase (AGT)
7.6. Human Pyridoxine 5′-Phosphate Oxidase (PNPOx)
- Capturing reaction intermediates: Monitoring the structural changes of PNPOx and its active site residues during the catalytic cycle will allow us to elucidate the catalytic mechanism of PNPOx, including the proton transfer steps.
- Investigating transfer and channeling mechanism: By capturing the structural dynamics of PNPOx in complex with PLP and potential protein partners involved in PLP transfer, it will be possible to gain more insights into the mechanisms underlying substrate channeling and enzyme regulation.
- Identifying allosteric regulation: Understanding the allosteric regulation of PNPOx will be critical for deciphering its physiological roles and developing allosteric modulators as potential therapeutic agents.
8. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef] [PubMed]
- Radzicka, A.; Wolfenden, R. A Proficient Enzyme. Science 1995, 267, 90–93. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Rahman, S.; Keller, M.; Zschocke, J.; Abdenur, J.; Ali, H.; Artuch, R.; Ballabio, A.; Barshop, B.; Baumgartner, M.; et al. An international classification of inherited metabolic disorders (ICIMD). J. Inherit. Metab. Dis. 2021, 44, 164–177. [Google Scholar] [CrossRef]
- Eckel, R.H.; Alberti, K.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2010, 375, 181–183. [Google Scholar] [CrossRef]
- Sreedhar, A.; Zhao, Y. Dysregulated metabolic enzymes and metabolic reprogramming in cancer cells. Biomed. Rep. 2018, 8, 3–10. [Google Scholar] [CrossRef]
- Elabbassi, W.N.; Haddad, H.A. The epidemic of the metabolic syndrome. Saudi Med. J. 2005, 26, 373–375. [Google Scholar] [PubMed]
- Chew, N.W.S.; Ng, C.H.; Tan, D.J.H.; Kong, G.; Lin, C.; Chin, Y.H.; Lim, W.H.; Huang, D.Q.; Quek, J.; Fu, C.E.; et al. The global burden of metabolic disease: Data from 2000 to 2019. Cell Metab. 2023, 35, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Wolynes, P.G. A second molecular biology revolution? the energy landscapes of biomolecular function. Phys. Chem. Chem. Phys. 2014, 16, 6321–6322. [Google Scholar] [CrossRef]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef]
- Wendt, K.U.; Weiss, M.S.; Cramer, P.; Heinz, D.W. Structures and diseases. Nat. Struct. Mol. Biol. 2008, 15, 117–120. [Google Scholar] [CrossRef]
- Agarwal, P.K.; Bernard, D.N.; Bafna, K.; Doucet, N. Enzyme Dynamics: Looking Beyond a Single Structure. ChemCatChem 2020, 12, 4704–4720. [Google Scholar] [CrossRef]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. A practical guide to modelling enzyme-catalysed reactions. Chem. Soc. Rev. 2012, 41, 3025–3038. [Google Scholar] [CrossRef]
- de Chadarevian, S. John Kendrew and myoglobin: Protein structure determination in the 1950s. Protein Sci. 2018, 27, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.W. From structural biology to designing therapy for inborn errors of metabolism. J. Inherit. Metab. Dis. 2016, 39, 489–498. [Google Scholar] [CrossRef]
- Kang, T.S.; Stevens, R.C. Structural aspects of therapeutic enzymes to treat metabolic disorders. Hum. Mutat. 2009, 30, 1591–1610. [Google Scholar] [CrossRef]
- Medina-Carmona, E.; Betancor-Fernández, I.; Santos, J.; Mesa-Torres, N.; Grottelli, S.; Batlle, C.; Naganathan, A.N.; Oppici, E.; Cellini, B.; Ventura, S.; et al. Insight into the specificity and severity of pathogenic mechanisms associated with missense mutations through experimental and structural perturbation analyses. Hum. Mol. Genet. 2019, 28, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Berman, H.M.; Duarte, J.M.; Feng, Z.; Flatt, J.W.; Hudson, B.P.; Lowe, R.; Peisach, E.; Piehl, D.W.; Rose, Y.; et al. Protein Data Bank: A Comprehensive Review of 3D Structure Holdings and Worldwide Utilization by Researchers, Educators, and Students. Biomolecules 2022, 12, 1425. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, A.C. X-ray Crystallography in Structure—Function Characterization of Therapeutic Enzymes BT—Therapeutic Enzymes: Function and Clinical Implications; Labrou, N., Ed.; Springer: Singapore, 2019; pp. 81–103. [Google Scholar]
- Smyth, M.S.; Martin, J.H.J. X-ray crystallography. Mol. Pathol. 2000, 53, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Donkor, A.K.; Pagare, P.P.; Mughram, M.H.A.L.; Safo, M.K. X-ray crystallography and sickle cell disease drug discovery—A tribute to Donald Abraham. Front. Mol. Biosci. 2023, 10, 1136970. [Google Scholar] [CrossRef]
- Zheng, H.; Handing, K.B.; Zimmerman, M.D.; Shabalin, I.G.; Almo, S.C.; Minor, W. X-ray crystallography over the past decade for novel drug discovery—Where are we heading next? Expert Opin. Drug Discov. 2015, 10, 975–989. [Google Scholar] [CrossRef]
- Maveyraud, L.; Mourey, L. Protein X-ray crystallography and drug discovery. Molecules 2020, 25, 5. [Google Scholar] [CrossRef] [PubMed]
- Rathore, I.; Mishra, V.; Bhaumik, P. Advancements in macromolecular crystallography: From past to present. Emerg. Top. Life Sci. 2021, 5, 127–149. [Google Scholar] [PubMed]
- Schmidt, M. Macromolecular movies, storybooks written by nature. Biophys. Rev. 2021, 13, 1191–1197. [Google Scholar] [CrossRef]
- Dunge, A.; Phan, C.; Uwangue, O.; Bjelcic, M.; Gunnarsson, J.; Wehlander, G.; Käck, H.; Brändén, G. Exploring serial crystallography for drug discovery. IUCrJ 2024, 11. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M. Time-Resolved Macromolecular Crystallography at Pulsed X-ray Sources. Int. J. Mol. Sci. 2019, 20, 1401. [Google Scholar] [CrossRef]
- Taberman, H. Radiation damage in macromolecular crystallography—An experimentalist’s view. Crystals 2018, 8, 157. [Google Scholar] [CrossRef]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; DePonte, D.P.; Weierstall, U.; et al. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–77. [Google Scholar] [CrossRef]
- Martin-Garcia, J.M.; Conrad, C.E.; Coe, J.; Roy-Chowdhury, S.; Fromme, P. Serial femtosecond crystallography: A revolution in structural biology. Arch. Biochem. Biophys. 2016, 602, 32–47. [Google Scholar] [CrossRef]
- Doscher, M.S.; Richards, F.M. The Activity of an Enzyme in the Crystalline State: Ribonuclease S. J. Biol. Chem. 1963, 238, 2399–2406. [Google Scholar] [CrossRef]
- Forneris, F.; Burnley, B.T.; Gros, P. Ensemble refinement shows conformational flexibility in crystal structures of human complement factor D. Acta Crystallogr. Sect. D 2014, 70, 733–743. [Google Scholar] [CrossRef]
- Tyka, M.D.; Keedy, D.A.; André, I.; DiMaio, F.; Song, Y.; Richardson, D.C.; Richardson, J.S.; Baker, D. Alternate States of Proteins Revealed by Detailed Energy Landscape Mapping. J. Mol. Biol. 2011, 405, 607–618. [Google Scholar] [CrossRef]
- Hekstra, D.R.; White, K.I.; Socolich, M.A.; Henning, R.W.; Šrajer, V.; Ranganathan, R. Electric-field-stimulated protein mechanics. Nature 2016, 540, 400–405. [Google Scholar] [CrossRef]
- Keedy, D.A.; Kenner, L.R.; Warkentin, M.; Woldeyes, R.A.; Hopkins, J.B.; Thompson, M.C.; Brewster, A.S.; Van Benschoten, A.H.; Baxter, E.L.; Uervirojnangkoorn, M.; et al. Mapping the conformational landscape of a dynamic enzyme by multitemperature and XFEL crystallography. eLife 2015, 4, 1–26. [Google Scholar] [CrossRef]
- Davidi, D.; Longo, L.M.; Jabłońska, J.; Milo, R.; Tawfik, D.S. A Bird’s-Eye View of Enzyme Evolution: Chemical, Physicochemical, and Physiological Considerations. Chem. Rev. 2018, 118, 8786–8797. [Google Scholar] [CrossRef] [PubMed]
- Jeske, L.; Placzek, S.; Schomburg, I.; Chang, A.; Schomburg, D. BRENDA in 2019: A European ELIXIR core data resource. Nucleic Acids Res. 2019, 47, 542–549. [Google Scholar] [CrossRef]
- Walsh, C. Enabling the chemistry of life. Nature 2001, 409, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Benkovic, S.J.; Hammes-Schiffer, S. A Perspective on Enzyme Catalysis. Science 2003, 301, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A.; Bora, R.P. Perspective: Defining and quantifying the role of dynamics in enzyme catalysis. J. Chem. Phys. 2016, 144, 180901. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.A.; Lei, M.; Thai, V.; Kerns, S.J.; Karplus, M.; Kern, D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 180901. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M. Reaction initiation in enzyme crystals by diffusion of substrate. Crystals 2020, 10, 116. [Google Scholar] [CrossRef]
- Schmidt, M. Mix and Inject: Reaction Initiation by Diffusion for Time-Resolved Macromolecular Crystallography. Adv. Condens. Matter Phys. 2013, 2013, 167276. [Google Scholar] [CrossRef]
- Emma, P.; Akre, R.; Arthur, J.; Bionta, R.; Bostedt, C.; Bozek, J.; Brachmann, A.; Bucksbaum, P.; Coffee, R.; Decker, F.-J.; et al. First lasing and operation of an Angstrom-wavelength free-electron laser. Nat. Photonics 2010, 4, 641–647. [Google Scholar] [CrossRef]
- Neutze, R.; Wouts, R.; Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 2000, 406, 6. [Google Scholar] [CrossRef]
- Echelmeier, A.; Sonker, M.; Ros, A. Microfluidic sample delivery for serial crystallography using XFELs. Anal. Bioanal. Chem. 2019, 411, 6535–6547. [Google Scholar] [CrossRef]
- White, T.A.; Mariani, V.; Brehm, W.; Yefanov, O.; Barty, A.; Beyerlein, K.R.; Chervinskii, F.; Galli, L.; Gati, C.; Nakane, T.; et al. Recent developments in CrystFEL. J. Appl. Crystallogr. 2016, 49, 680–689. [Google Scholar] [CrossRef]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 85–97. [Google Scholar] [CrossRef]
- Fromme, P.; Graves, W.S.; Martin-Garcia, J.M. Serial Femtosecond Crystallography: A Decade at the Forefront in Structural Biology. eLS 2020, 1–17. [Google Scholar] [CrossRef]
- Malla, T.N.; Schmidt, M. Transient state measurements on proteins by time-resolved crystallography. Curr. Opin. Struct. Biol. 2022, 74, 102376. [Google Scholar] [CrossRef]
- Martin-Garcia, J.M. Protein dynamics and time resolved protein crystallography at synchrotron radiation sources: Past, present and future. Crystals 2021, 11, 521. [Google Scholar] [CrossRef]
- Stagno, J.R.; Liu, Y.; Bhandari, Y.R.; Conrad, C.E.; Panja, S.; Swain, M.; Fan, L.; Nelson, G.; Li, C.; Wendel, D.R.; et al. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature 2017, 541, 242–246. [Google Scholar] [CrossRef]
- Kupitz, C.; Olmos, J.L.; Holl, M.; Tremblay, L.; Pande, L.; Pandey, S.; Oberthür, D.; Hunter, M.; Liang, M.; Aquila, A.; et al. Structural enzymology using X-ray free electron lasers. Struct. Dyn. 2017, 4, 044003. [Google Scholar] [CrossRef] [PubMed]
- Olmos, J.L.; Pandey, S.; Martin-Garcia, J.M.; Calvey, G.; Katz, A.; Knoska, J.; Kupitz, C.; Hunter, M.S.; Liang, M.; Oberthuer, D.; et al. Enzyme intermediates captured ‘on the fly’ by mix-and-inject serial crystallography. BMC Biol. 2018, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Calvey, G.; Katz, A.M.; Malla, T.N.; Koua, F.H.M.; Martin-Garcia, J.M.; Poudyal, I.; Yang, J.H.; Vakili, M.; Yefanov, O.; et al. Observation of substrate diffusion and ligand binding in enzyme crystals using high-repetition-rate mix-and-inject serial crystallography. IUCrJ 2021, 8, 878–895. [Google Scholar] [CrossRef] [PubMed]
- Grieco, A.; Boneta, S.; Gavira, J.A.; Pey, A.L.; Basu, S.; Orlans, J.; de Sanctis, D.; Medina, M.; Martin-Garcia, J.M. Structural dynamics and functional cooperativity of human NQO1 by ambient temperature serial crystallography and simulations. Protein Sci. 2024, 33, e4957. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.C.F.; Koenig, D.F.; Mair, G.A.; North, A.C.T.; Phillips, D.C.; Sarma, V.R. Structure of Hen Egg-White Lysozyme: A Three-dimensional Fourier Synthesis at 2 Å Resolution. Nature 1965, 206, 757–761. [Google Scholar] [CrossRef]
- Wilson, M.A. Mapping Enzyme Landscapes by Time-Resolved Crystallography with Synchrotron and X-ray Free Electron Laser Light. Annu. Rev. Biophys. 2022, 51, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Hekstra, D.R. Emerging Time-Resolved X-ray Diffraction Approaches for Protein Dynamics. Annu. Rev. Biophys. 2023, 52, 255–274. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef]
- Bhabha, G.; Biel, J.T.; Fraser, J.S. Keep on moving: Discovering and perturbing the conformational dynamics of enzymes. Acc. Chem. Res. 2015, 48, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bedem, H.; Fraser, J.S. Integrative, dynamic structural biology at atomic resolution—It’s about time. Nat. Methods 2015, 12, 307–318. [Google Scholar] [CrossRef]
- Coquelle, N.; Sliwa, M.; Woodhouse, J.; Schirò, G.; Adam, V.; Aquila, A.; Barends, T.R.M.; Boutet, S.; Byrdin, M.; Carbajo, S.; et al. Chromophore twisting in the excited state of a photoswitchable fluorescent protein captured by time-resolved serial femtosecond crystallography. Nat. Chem. 2018, 10, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Genick, U.K.; Borgstahl, G.E.O.; Ng, K.; Ren, Z.; Pradervand, C.; Burke, P.M.; Šrajer, V.; Teng, T.-Y.; Schildkamp, W.; McRee, D.E.; et al. Structure of a Protein Photocycle Intermediate by Millisecond Time-Resolved Crystallography. Science 1997, 275, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Schlichting, I.; Almo, S.C.; Rapp, G.; Wilson, K.; Petratos, K.; Lentfer, A.; Wittinghofer, A.; Kabsch, W.; Pai, E.F.; Petsko, G.A.; et al. Time–resolved X-ray crystallographic study of the conformational change in Ha–Ras p21 protein on GTP hydrolysis. Nature 1990, 345, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Šrajer, V.; Teng, T.; Ursby, T.; Pradervand, C.; Ren, Z.; Adachi, S.; Schildkamp, W.; Bourgeois, D.; Wulff, M.; Moffat, K. Photolysis of the Carbon Monoxide Complex of Myoglobin: Nanosecond Time-Resolved Crystallography. Science 1996, 274, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Neutze, R.; Moffat, K. Time-resolved structural studies at synchrotrons and X-ray free electron lasers: Opportunities and challenges. Curr. Opin. Struct. Biol. 2012, 22, 651–659. [Google Scholar] [CrossRef]
- Orville, A.M. Recent results in time resolved serial femtosecond crystallography at XFELs. Curr. Opin. Struct. Biol. 2020, 65, 193–208. [Google Scholar] [CrossRef]
- Schmidt, M. Time-Resolved Macromolecular Crystallography at Modern X-ray Sources BT—Protein Crystallography: Methods and Protocols; Wlodawer, A., Dauter, Z., Jaskolski, M., Eds.; Springer: New York, NY, USA, 2017; pp. 273–294. [Google Scholar]
- Josts, I.; Niebling, S.; Gao, Y.; Levantino, M.; Tidow, H.; Monteiro, D. Photocage-initiated time-resolved solution X-ray scattering investigation of protein dimerization. IUCrJ 2018, 5, 667–672. [Google Scholar] [CrossRef]
- Dasgupta, M.; Budday, D.; de Oliveira, S.H.P.; Madzelan, P.; Marchany-Rivera, D.; Seravalli, J.; Hayes, B.; Sierra, R.G.; Boutet, S.; Hunter, M.S.; et al. Mix-and-inject XFEL crystallography reveals gated conformational dynamics during enzyme catalysis. Proc. Natl. Acad. Sci. USA 2019, 116, 25634–25640. [Google Scholar] [CrossRef]
- Ishigami, I.; Lewis-Ballester, A.; Echelmeier, A.; Brehm, G.; Zatsepin, N.A.; Grant, T.D.; Coe, J.D.; Lisova, S.; Nelson, G.; Zhang, S.; et al. Snapshot of an oxygen intermediate in the catalytic reaction of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2019, 116, 3572–3577. [Google Scholar] [CrossRef]
- Mehrabi, P.; Schulz, E.C.; Agthe, M.; Horrell, S.; Bourenkov, G.; von Stetten, D.; Leimkohl, J.-P.; Schikora, H.; Schneider, T.R.; Pearson, A.R.; et al. Liquid application method for time-resolved analyses by serial synchrotron crystallography. Nat. Methods 2019, 16, 979–982. [Google Scholar] [CrossRef]
- Calvey, G.D.; Katz, A.M.; Pollack, L. Microfluidic Mixing Injector Holder Enables Routine Structural Enzymology Measurements with Mix-and-Inject Serial Crystallography Using X-ray Free Electron Lasers. Anal. Chem. 2019, 91, 7139–7144. [Google Scholar] [CrossRef]
- Murakawa, T.; Suzuki, M.; Fukui, K.; Masuda, T.; Sugahara, M.; Tono, K.; Tanaka, T.; Iwata, S.; Nango, E.; Yano, T.; et al. Serial femtosecond X-ray crystallography of an anaerobically formed catalytic intermediate of copper amine oxidase. Acta Crystallogr. Sect. D Struct. Biol. 2022, 78, 1428–1438. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. Functions of NQO1 in cellular protection and CoQ10 metabolism and its potential role as a redox sensitive molecular switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef]
- Gaikwad, A.; Long, D.J.; Stringer, J.L.; Jaiswal, A.K. In Vivo Role of NAD(P)H:Quinone Oxidoreductase 1 (NQO1) in the Regulation of Intracellular Redox State and Accumulation of Abdominal Adipose Tissue. J. Biol. Chem. 2001, 276, 22559–22564. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef]
- Siegel, D.; Ross, D. Immunodetection of NAD(P)H:quinone oxidoreductase 1 (NQO1) in human tissues. Free Radic. Biol. Med. 2000, 29, 246–253. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar] [CrossRef]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The crystal structure of NAD(P)H quinone oxidoreductase 1 in complex with its potent inhibitor dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef]
- Faig, M.; Bianchet, M.A.; Talalay, P.; Chen, S.; Winski, S.; Ross, D.; Amzel, L.M. Structures of recombinant human and mouse NAD(P)H:quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. USA 2000, 97, 3177–3182. [Google Scholar] [CrossRef]
- Li, R.; Bianchet, M.A.; Talalay, P.; Amzel, L.M. The three-dimensional structure of NAD(P)H:quinone reductase a flavoprotein involved in cancer chemoprotection and chemotherapy: Mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. USA 1995, 92, 8846–8850. [Google Scholar] [CrossRef]
- Skelly, J.V.; Sanderson, M.R.; Suter, D.A.; Baumann, U.; Read, M.A.; Gregory, D.S.J.; Bennett, M.; Hobbs, S.M.; Neidle, S. Crystal structure of human DT-diaphorase: A model for interaction with the cytotoxic prodrug 5-(aziridine-1-yl)-2,4-dinitrobenzamide (CB1954). J. Med. Chem. 1999, 42, 4325–4330. [Google Scholar] [CrossRef] [PubMed]
- Onyenwoke, R.U.; Wiegel, J. Iron (III) reduction: A novel activity of the human NAD(P)H:oxidoreductase. Biochem. Biophys. Res. Commun. 2007, 353, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Anusevičius, Ž.; Šarlauskas, J.; Čenas, N. Two-electron reduction of quinones by rat liver NAD(P)H:quinone oxidoreductase: Quantitative structure-activity relationships. Arch. Biochem. Biophys. 2002, 404, 254–262. [Google Scholar] [CrossRef]
- Anoz-Carbonell, E.; Timson, D.J.; Pey, A.L.; Medina, M. The catalytic cycle of the antioxidant and cancer-associated human NQO1 enzyme: Hydride transfer, conformational dynamics and functional cooperativity. Antioxidants 2020, 9, 772. [Google Scholar] [CrossRef]
- Lienhart, W.D.; Gudipati, V.; Uhl, M.K.; Binter, A.; Pulido, S.A.; Saf, R.; Zangger, K.; Gruber, K.; Macheroux, P. Collapse of the native structure caused by a single amino acid exchange in human NAD(P)H:Quinone oxidoreductase. FEBS J. 2014, 281, 4691–4704. [Google Scholar] [CrossRef]
- Winski, S.L.; Faig, M.; Bianchet, M.A.; Siegel, D.; Swann, E.; Fung, K.; Duncan, M.W.; Moody, C.J.; Amzel, L.M.; Ross, D. Characterization of a mechanism-based inhibitor of NAD(P)H:Quinone oxidoreductase 1 by biochemical, X-ray crystallographic, and mass spectrometric approaches. Biochemistry 2001, 40, 15135–15142. [Google Scholar] [CrossRef]
- Doppler, D.; Sonker, M.; Egatz-gomez, A.; Grieco, A.; Zaare, S.; Jernigan, R.; Meza-aguilar, J.D.; Rabbani, M.T.; Manna, A.; Alvarez, R.C.; et al. Modular droplet injector for sample conservation conformational heterogeneity in the disease-associated NQO1 enzyme. Lab Chip 2023, 23, 3016–3033. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef]
- Milano, S.K.; Huang, Q.; Nguyen, T.T.T.; Ramachandran, S.; Finke, A.; Kriksunov, I.; Schuller, D.J.; Szebenyi, D.M.; Arenholz, E.; McDermott, L.A.; et al. New insights into the molecular mechanisms of glutaminase C inhibitors in cancer cells using serial room temperature crystallography. J. Biol. Chem. 2022, 298, 101535. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Stalnecker, C.; Zhang, C.; McDermott, L.A.; Iyer, P.; O’Neill, J.; Reimer, S.; Cerione, R.A.; Katt, W.P. Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism. J. Biol. Chem. 2018, 293, 3535–3545. [Google Scholar] [CrossRef]
- Nguyen, R.C.; Davis, I.; Dasgupta, M.; Wang, Y.; Simon, P.S.; Butryn, A.; Makita, H.; Bogacz, I.; Dornevil, K.; Aller, P.; et al. In Situ Structural Observation of a Substrate- and Peroxide-Bound High-Spin Ferric-Hydroperoxo Intermediate in the P450 Enzyme CYP121. J. Am. Chem. Soc. 2023, 145, 25120–25133. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, T.; Lu, X.; Lan, X.; Chen, Z.; Lu, S. G protein-coupled receptors (GPCRs): Advances in structures, mechanisms, and drug discovery. Signal Transduct. Target. Ther. 2024, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Heng, B.C.; Aubel, D.; Fussenegger, M. An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol. Adv. 2013, 31, 1676–1694. [Google Scholar] [CrossRef]
- Wong, T.-S.; Li, G.; Li, S.; Gao, W.; Chen, G.; Gan, S.; Zhang, M.; Li, H.; Wu, S.; Du, Y. G protein-coupled receptors in neurodegenerative diseases and psychiatric disorders. Signal Transduct. Target. Ther. 2023, 8, 177. [Google Scholar]
- Li, Y.; Li, B.; Chen, W.-D.; Wang, Y.-D. Role of G-protein coupled receptors in cardiovascular diseases. Front. Cardiovasc. Med. 2023, 10, 1130312. [Google Scholar] [CrossRef]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G protein-coupled receptors: Structure- and function-based drug discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef]
- Liu, W.; Wacker, D.; Gati, C.; Han, G.W.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; Katritch, V.; Barty, A.; et al. Serial Femtosecond Crystallography of G Protein-Coupled Receptors. Science 2013, 342, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Han, G.W.; Batyuk, A.; Ishchenko, A.; White, K.L.; Patel, N.; Sadybekov, A.; Zamlynny, B.; Rudd, M.T.; Hollenstein, K.; et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nature 2017, 544, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Audet, M.; White, K.L.; Breton, B.; Zarzycka, B.; Han, G.W.; Lu, Y.; Gati, C.; Batyuk, A.; Popov, P.; Velasquez, J.; et al. Crystal structure of misoprostol bound to the labor inducer prostaglandin E2 receptor. Nat. Chem. Biol. 2019, 15, 11–17. [Google Scholar] [CrossRef]
- Johansson, L.C.; Stauch, B.; McCorvy, J.D.; Han, G.W.; Patel, N.; Huang, X.P.; Batyuk, A.; Gati, C.; Slocum, S.T.; Li, C.; et al. XFEL structures of the human MT2 melatonin receptor reveal the basis of subtype selectivity. Nature 2019, 569, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef]
- Ishchenko, A.; Wacker, D.; Kapoor, M.; Zhang, A.; Han, G.W.; Basu, S.; Patel, N.; Messerschmidt, M.; Weierstall, U.; Liu, W.; et al. Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody. Proc. Natl. Acad. Sci. USA 2017, 114, 8223–8228. [Google Scholar] [CrossRef]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 2017, 170, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qiao, A.; Yang, D.; Yang, L.; Dai, A.; de Graaf, C.; Reedtz-Runge, S.; Dharmarajan, V.; Zhang, H.; Han, G.W.; et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 2017, 546, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, F.; Wu, Y.; Yang, J.; Han, G.W.; Zhao, S.; Ishchenko, A.; Ye, L.; Lin, X.; Ding, K.; et al. Crystal structure of a multi-domain human smoothened receptor in complex with a super stabilizing ligand. Nat. Commun. 2017, 8, 15383. [Google Scholar] [CrossRef]
- Johansson, L.C.; Stauch, B.; Ishchenko, A.; Cherezov, V. A Bright Future for Serial Femtosecond Crystallography with XFELs. Trends Biochem. Sci. 2017, 42, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Stauch, B.; Cherezov, V. Serial Femtosecond Crystallography of G Protein-Coupled Receptors. Annu. Rev. Biophys. 2018, 47, 377–397. [Google Scholar] [CrossRef] [PubMed]
- Mishin, A.; Gusach, A.; Luginina, A.; Marin, E.; Borshchevskiy, V.; Cherezov, V. An outlook on using serial femtosecond crystallography in drug discovery. Expert Opin. Drug Discov. 2019, 14, 933–945. [Google Scholar] [CrossRef]
- Hemschemeier, A.; Happe, T. The plasticity of redox cofactors: From metalloenzymes to redox-active DNA. Nat. Rev. Chem. 2018, 2, 231–243. [Google Scholar] [CrossRef]
- Nastri, F.; D’Alonzo, D.; Leone, L.; Zambrano, G.; Pavone, V.; Lombardi, A. Engineering Metalloprotein Functions in Designed and Native Scaffolds. Trends Biochem. Sci. 2019, 44, 1022–1040. [Google Scholar] [CrossRef] [PubMed]
- Waldron, K.J.; Rutherford, J.C.; Ford, D.; Robinson, N.J. Metalloproteins and metal sensing. Nature 2009, 460, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Prabhulkar, S.; Tian, H.; Wang, X.; Zhu, J.-J.; Li, C.-Z. Engineered Proteins: Redox Properties and Their Applications. Antioxid. Redox Signal. 2012, 17, 1796–1822. [Google Scholar] [CrossRef]
- Sigfridsson, K.G.V.; Chernev, P.; Leidel, N.; Popović-Bijelić, A.; Gräslund, A.; Haumann, M. Rapid X-ray photoreduction of dimetal-oxygen cofactors in ribonucleotide reductase. J. Biol. Chem. 2013, 288, 9648–9661. [Google Scholar] [CrossRef]
- Yano, J.; Kern, J.; Irrgang, K.-D.; Latimer, M.J.; Bergmann, U.; Glatzel, P.; Pushkar, Y.; Biesiadka, J.; Loll, B.; Sauer, K.; et al. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: A case study for metalloprotein crystallography. Proc. Natl. Acad. Sci. USA 2005, 102, 12047–12052. [Google Scholar] [CrossRef]
- Garman, E.F.; Weik, M. Radiation damage to macromolecules: Kill or cure? J. Synchrotron Radiat. 2015, 22, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Nass, K. Radiation damage in protein crystallography at X-ray free-electron lasers. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 211–218. [Google Scholar] [CrossRef]
- Kern, J.; Chatterjee, R.; Young, I.D.; Fuller, F.D.; Lassalle, L.; Ibrahim, M.; Gul, S.; Fransson, T.; Brewster, A.S.; Alonso-Mori, R.; et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425. [Google Scholar] [CrossRef]
- Kern, J.; Tran, R.; Alonso-Mori, R.; Koroidov, S.; Echols, N.; Hattne, J.; Ibrahim, M.; Gul, S.; Laksmono, H.; Sierra, R.G.; et al. Taking snapshots of photosynthetic water oxidation using femtosecond X-ray diffraction and spectroscopy. Nat. Commun. 2014, 5, 4371. [Google Scholar] [CrossRef]
- Sauter, N.K.; Kern, J.; Yano, J.; Holton, J.M. Towards the spatial resolution of metalloprotein charge states by detailed modeling of XFEL crystallographic diffraction. Acta Crystallogr. Sect. D Struct. Biol. 2020, 76, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Suga, M.; Shimada, A.; Akita, F.; Shen, J.R.; Tosha, T.; Sugimoto, H. Time-resolved studies of metalloproteins using X-ray free electron laser radiation at SACLA. Biochim. Biophys. Acta—Gen. Subj. 2020, 1864, 129466. [Google Scholar] [CrossRef]
- Lancaster, K.M.; Roemelt, M.; Ettenhuber, P.; Hu, Y.; Ribbe, M.W.; Neese, F.; Bergmann, U.; DeBeer, S. X-ray emission spectroscopy evidences a central carbon in the nitrogenase iron-molybdenum cofactor. Science 2011, 334, 974–977. [Google Scholar] [CrossRef]
- Lomelino, C.L.; Kim, J.K.; Lee, C.; Lim, S.W.; Andring, J.T.; Mahon, B.P.; Chung, M.; Kim, C.U.; McKenna, R. Carbonic anhydrase II microcrystals suitable for XFEL studies. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74, 327–330. [Google Scholar] [CrossRef]
- Flydal, M.I.; Martinez, A. Phenylalanine hydroxylase: Function, structure, and regulation. IUBMB Life 2013, 65, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tong, X.; Zhang, S.; Wang, D.; Wang, L.; Wang, Q.; Fan, H. The Roles of Dipeptidyl Peptidase 4 (DPP4) and DPP4 Inhibitors in Different Lung Diseases: New Evidence. Front. Pharmacol. 2021, 12, 731453. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.; Price, A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar] [PubMed]
- Michalets, E.L. Update: Clinically Significant Cytochrome P-450 Drug Interactions. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1998, 18, 84–112. [Google Scholar] [CrossRef]
- Dong, A.N.; Ahemad, N.; Pan, Y.; Palanisamy, U.D.; Yiap, B.C.; Ong, C.E. Functional and structural characterisation of common cytochrome P450 2D6 allelic variants—Roles of Pro34 and Thr107 in catalysis and inhibition. Naunyn. Schmiedebergs. Arch. Pharmacol. 2019, 392, 1015–1029. [Google Scholar] [CrossRef]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: The past, present and future. Trends Pharmacol. Sci. 2004, 25, 193–200. [Google Scholar] [CrossRef]
- Hayaishi, O. An odyssey with oxygen. Biochem. Biophys. Res. Commun. 2005, 338, 2–6. [Google Scholar] [CrossRef]
- Waterman, M.R. Professor Howard Mason and oxygen activation. Biochem. Biophys. Res. Commun. 2005, 338, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Salido, E.; Pey, A.L.; Rodriguez, R.; Lorenzo, V. Primary hyperoxalurias: Disorders of glyoxylate detoxification. Biochim. Biophys. Acta—Mol. Basis Dis. 2012, 1822, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Cellini, B.; Bertoldi, M.; Montioli, R.; Paiardini, A.; Borri Voltattorni, C. Human wild–type alanine:glyoxylate aminotransferase and its naturally occurring G82E variant: Functional properties and physiological implications. Biochem. J. 2007, 408, 39–50. [Google Scholar] [CrossRef]
- Hoppe, B.; Beck, B.B.; Milliner, D.S. The primary hyperoxalurias. Kidney Int. 2009, 75, 1264–1271. [Google Scholar] [CrossRef]
- Cochat, P.; Liutkus, A.; Fargue, S.; Basmaison, O.; Ranchin, B.; Rolland, M.-O. Primary hyperoxaluria type 1: Still challenging! Pediatr. Nephrol. 2006, 21, 1075–1081. [Google Scholar] [CrossRef]
- Lorenzo, V.; Alvarez, A.; Torres, A.; Torregrosa, V.; Hernández, D.; Salido, E. Presentation and role of transplantation in adult patients with type 1 primary hyperoxaluria and the I244T AGXT mutation: Single-center experience. Kidney Int. 2006, 70, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Cochat, P.; Fargue, S.; Harambat, J. Primary hyperoxaluria type 1: Strategy for organ transplantation. Curr. Opin. Organ Transplant. 2010, 15, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Roe, S.M.; Pearl, L.H.; Danpure, C.J. Crystallization and preliminary crystallographic analysis of human alanine:glyoxylate aminotransferase and its polymorphic variants. Acta Crystallogr. Sect. D 2001, 57, 1936–1937. [Google Scholar] [CrossRef]
- Zhang, X.; Roe, S.M.; Hou, Y.; Bartlam, M.; Rao, Z.; Pearl, L.H.; Danpure, C.J. Crystal structure of alanine:glyoxylate aminotransferase and the relationship between genotype and enzymatic phenotype in primary hyperoxaluria type 1. J. Mol. Biol. 2003, 331, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Danpure, C.J.; Fryer, P.; Griffiths, S.; Guttridge, K.M.; Jennings, P.R.; Allsop, J.; Moser, A.B.; Naidu, S.; Moser, H.W.; MacCollin, M.; et al. Cytosolic compartmentalization of hepatic alanine: Glyoxylate aminotransferase in patients with aberrant peroxisomal biogenesis and its effect on oxalate metabolism. J. Inherit. Metab. Dis. 1994, 17, 27–40. [Google Scholar] [CrossRef]
- Purdue, P.E.; Takada, Y.; Danpure, C.J. Identification of mutations associated with peroxisome-to-mitochondrion mistargeting of alanine/glyoxylate aminotransferase in primary hyperoxaluria type 1. J. Cell Biol. 1990, 111, 2341–2351. [Google Scholar] [CrossRef]
- Lumb, M.J.; Danpure, C.J. Functional synergism between the most common polymorphism in human alanine:glyoxylate aminotransferase and four of the most common disease-causing mutations. J. Biol. Chem. 2000, 275, 36415–36422. [Google Scholar] [CrossRef]
- Danpure, C.J.; Cooper, P.J.; Wise, P.J.; Jennings, P.R. An enzyme trafficking defect in two patients with primary hyperoxaluria type 1: Peroxisomal analine/glyoxylate aminotransferase rerouted to mitochondria. J. Cell Biol. 1989, 108, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Leiper, J.M.; Oatey, P.B.; Danpure, C.J. Inhibition of alanine:glyoxylate aminotransferase 1 dimerization is a prerequisite for its peroxisome-to-mitochondrion mistargeting in primary hyperoxaluria type 1. J. Cell Biol. 1996, 135, 939–951. [Google Scholar] [CrossRef]
- Lumb, M.J.; Drake, A.F.; Danpure, C.J. Effect of N-terminal α-helix formation on the dimerization and intracellular targeting of alanine:glyoxylate aminotransferase. J. Biol. Chem. 1999, 274, 20587–20596. [Google Scholar] [CrossRef]
- Danpure, C.J. Primary hyperoxaluria type 1: AGT mistargeting highlights the fundamental differences between the peroxisomal and mitochondrial protein import pathways. Biochim. Biophys. Acta—Mol. Cell Res. 2006, 1763, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Garrelfs, S.F.; Frishberg, Y.; Hulton, S.A.; Koren, M.J.; O’Riordan, W.D.; Cochat, P.; Deschênes, G.; Shasha-Lavsky, H.; Saland, J.M.; van’t Hoff, W.G.; et al. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N. Engl. J. Med. 2021, 384, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Forbes, T.A.; Brown, B.D.; Lai, C. Therapeutic RNA interference: A novel approach to the treatment of primary hyperoxaluria. Br. J. Clin. Pharmacol. 2022, 88, 2525–2538. [Google Scholar] [CrossRef] [PubMed]
- Vankova, P.; Pacheco-Garcia, J.L.; Loginov, D.S.; Gómez-Mulas, A.; Kádek, A.; Martín-Garcia, J.M.; Salido, E.; Man, P.; Pey, A.L. Insights into the pathogenesis of primary hyperoxaluria type I from the structural dynamics of alanine:glyoxylate aminotransferase variants. FEBS Lett. 2024, 598, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, D.L.; Al-Shekaili, H.H.; Tarailo-Graovac, M.; Wolf, N.I.; Ivy, A.S.; Demarest, S.; Roussel, Y.; Ciapaite, J.; Van Roermund, C.W.T.; Kernohan, K.D. PLPHP deficiency: Clinical, genetic, biochemical, and mechanistic insights. Brain 2019, 142, 542–559. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.P.; Plecko, B.; Mills, P.B.; Clayton, P.T. Disorders affecting vitamin B6 metabolism. J. Inherit. Metab. Dis. 2019, 42, 629–646. [Google Scholar] [CrossRef] [PubMed]
- ALMughram, M.H.; Ghatge, M.S.; Kellogg, G.E.; Safo, M.K. Elucidating the Interaction between Pyridoxine 5′-Phosphate Oxidase and Dopa Decarboxylase: Activation of B6-Dependent Enzyme. Int. J. Mol. Sci. 2023, 24, 642. [Google Scholar] [CrossRef]
- Ghatge, M.S.; Al Mughram, M.; Omar, A.M.; Safo, M.K. Inborn errors in the vitamin B6 salvage enzymes associated with neonatal epileptic encephalopathy and other pathologies. Biochimie 2021, 183, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Barile, A.; Mills, P.; Di Salvo, M.L.; Graziani, C.; Bunik, V.; Clayton, P.; Contestabile, R.; Tramonti, A. Characterization of novel pathogenic variants causing pyridox(Am)ine 5′–phosphate oxidase–dependent epilepsy. Int. J. Mol. Sci. 2021, 22, 12013. [Google Scholar] [CrossRef]
- Gdynia, H.-J.; Müller, T.; Sperfeld, A.-D.; Kühnlein, P.; Otto, M.; Kassubek, J.; Ludolph, A.C. Severe sensorimotor neuropathy after intake of highest dosages of vitamin B6. Neuromuscul. Disord. 2008, 18, 156–158. [Google Scholar] [CrossRef]
- Scott, K.; Zeris, S.; Kothari, M.J. Elevated B6 levels and peripheral neuropathies. Electromyogr. Clin. Neurophysiol. 2008, 48, 219–223. [Google Scholar]
- Foca, F.J. Motor and sensory neuropathy secondary to excessive pyridoxine ingestion. Arch. Phys. Med. Rehabil. 1985, 66, 634–636. [Google Scholar]
- Safo, M.K.; Mathews, I.; Musayev, F.N.; di Salvo, M.L.; Thiel, D.J.; Abraham, D.J.; Schirch, V. X-ray structure of Escherichia coli pyridoxine 5′-phosphate oxidase complexed with FMN at 1.8 A resolution. Structure 2000, 8, 751–762. [Google Scholar] [CrossRef]
- Di Salvo, M.L.; Ko, T.P.; Musayev, F.N.; Raboni, S.; Schirch, V.; Safo, M.K. Active site structure and stereospecificity of Escherichia coli pyridoxine-5′-phosphate oxidase. J. Mol. Biol. 2002, 315, 385–397. [Google Scholar] [CrossRef]
- Safo, M.K.; Musayev, F.N.; Di Salvo, M.L.; Schirch, V. X-ray structure of Escherichia coli pyridoxine 5′-phosphate oxidase complexed with pyridoxal 5′-phosphate at 2.0 Å resolution. J. Mol. Biol. 2001, 310, 817–826. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grieco, A.; Quereda-Moraleda, I.; Martin-Garcia, J.M. Innovative Strategies in X-ray Crystallography for Exploring Structural Dynamics and Reaction Mechanisms in Metabolic Disorders. J. Pers. Med. 2024, 14, 909. https://doi.org/10.3390/jpm14090909
Grieco A, Quereda-Moraleda I, Martin-Garcia JM. Innovative Strategies in X-ray Crystallography for Exploring Structural Dynamics and Reaction Mechanisms in Metabolic Disorders. Journal of Personalized Medicine. 2024; 14(9):909. https://doi.org/10.3390/jpm14090909
Chicago/Turabian StyleGrieco, Alice, Isabel Quereda-Moraleda, and Jose Manuel Martin-Garcia. 2024. "Innovative Strategies in X-ray Crystallography for Exploring Structural Dynamics and Reaction Mechanisms in Metabolic Disorders" Journal of Personalized Medicine 14, no. 9: 909. https://doi.org/10.3390/jpm14090909