
Co-Expression Network and Machine Learning Analysis of Transcriptomics Data Identifies Distinct Gene Signatures and Pathways in Lesional and Non-Lesional Atopic Dermatitis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Used in the Study
2.2. Differential Expression Genes (DEGs) Analysis
2.3. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.4. Identifying Modules Correlated with Lesional and Non-Lesional Skin
2.5. Functional Enrichment Analysis
2.6. Machine Learning Analysis of Pathway Expression and Statistical Analyses
3. Results
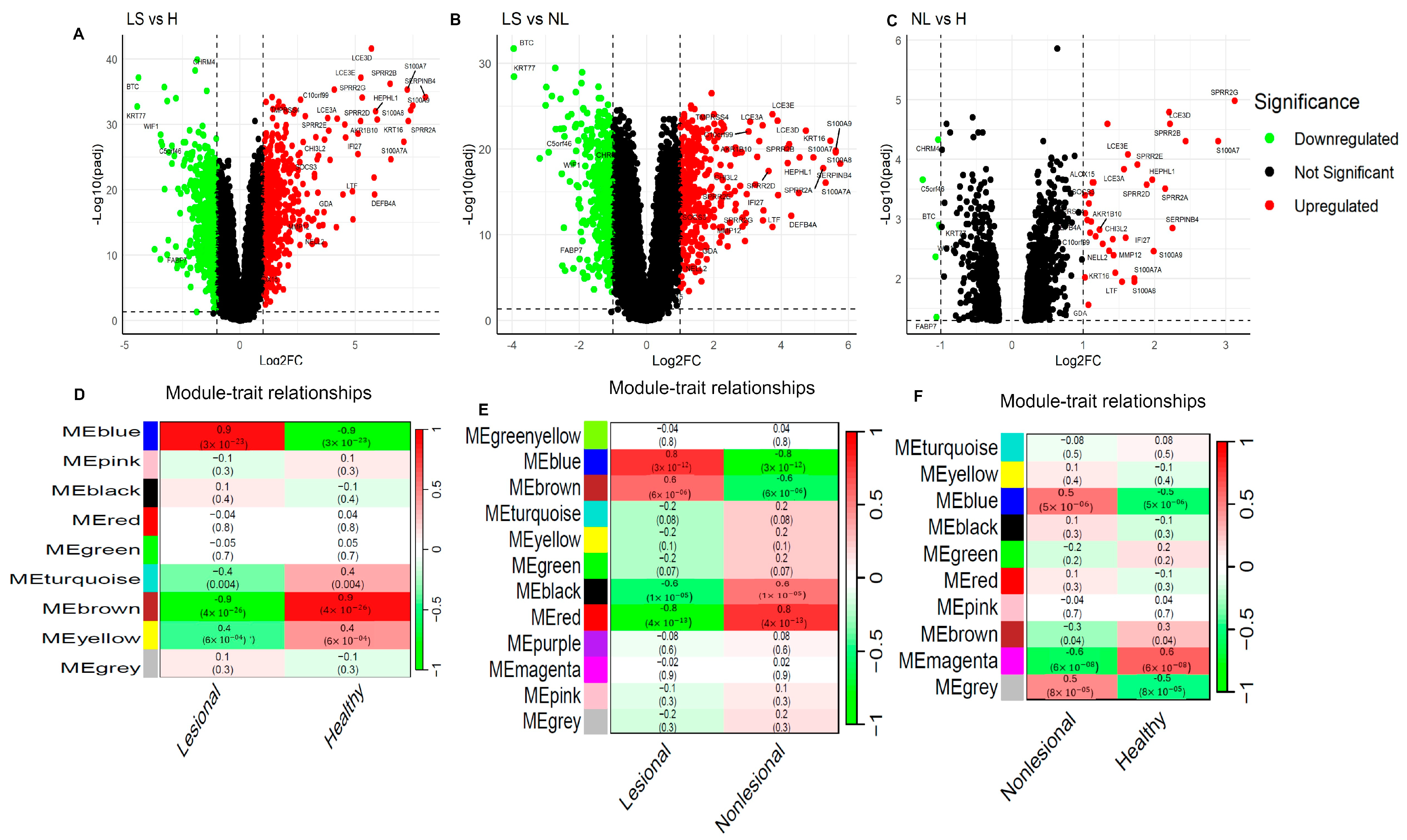
3.1. Identification of Differentially Expressed Genes in Lesional and Non-Lesional AD Skin
3.2. Identification of Differential Co-Expression Modules Associated with Lesional and Non-Lesional AD
3.3. Functional Enrichment Analysis of the DCEGs in Lesional and Non-Lesional Correlated Modules
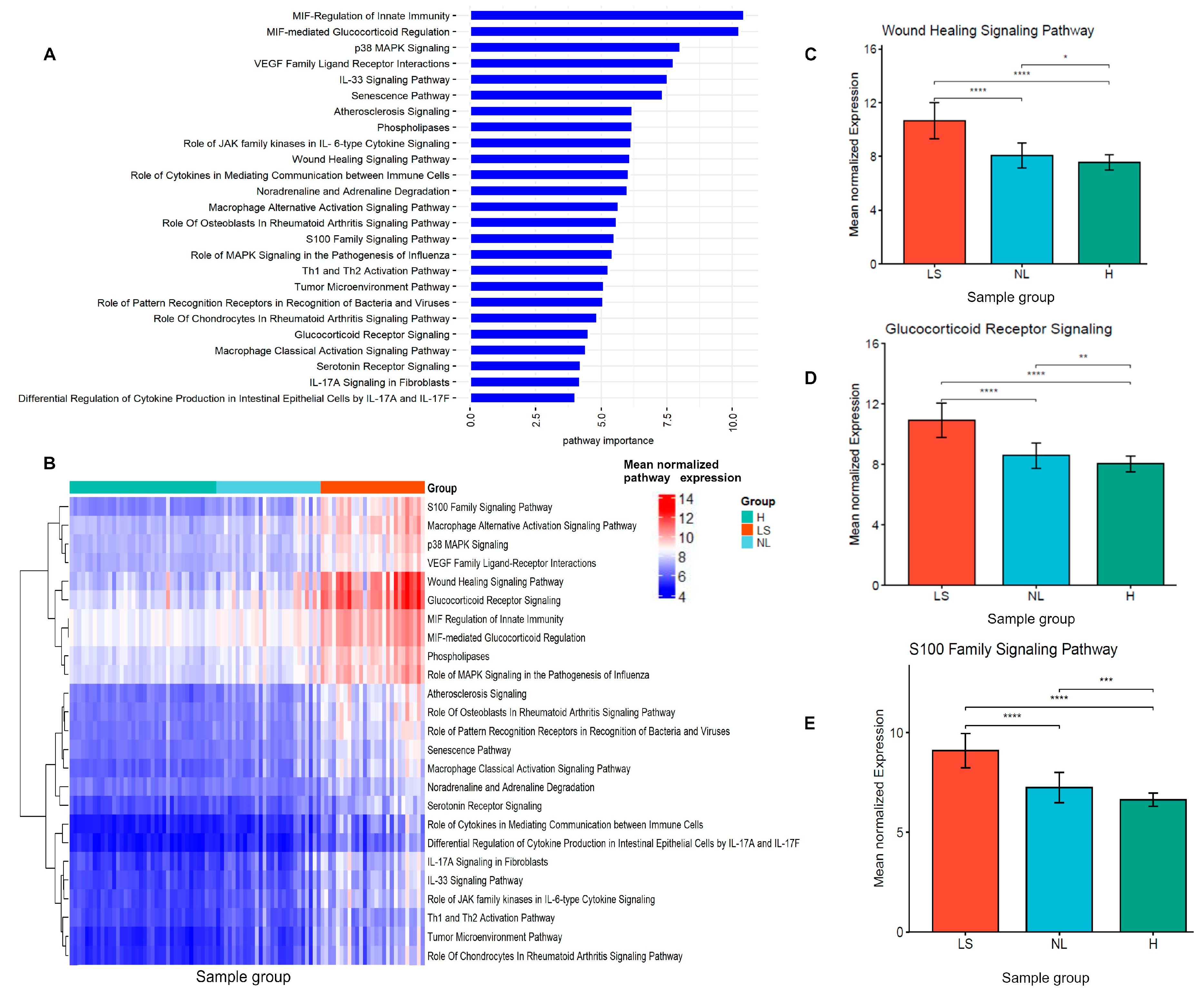
3.4. Identification of Lesional and Non-Lesional AD Discriminatory Pathway Signatures
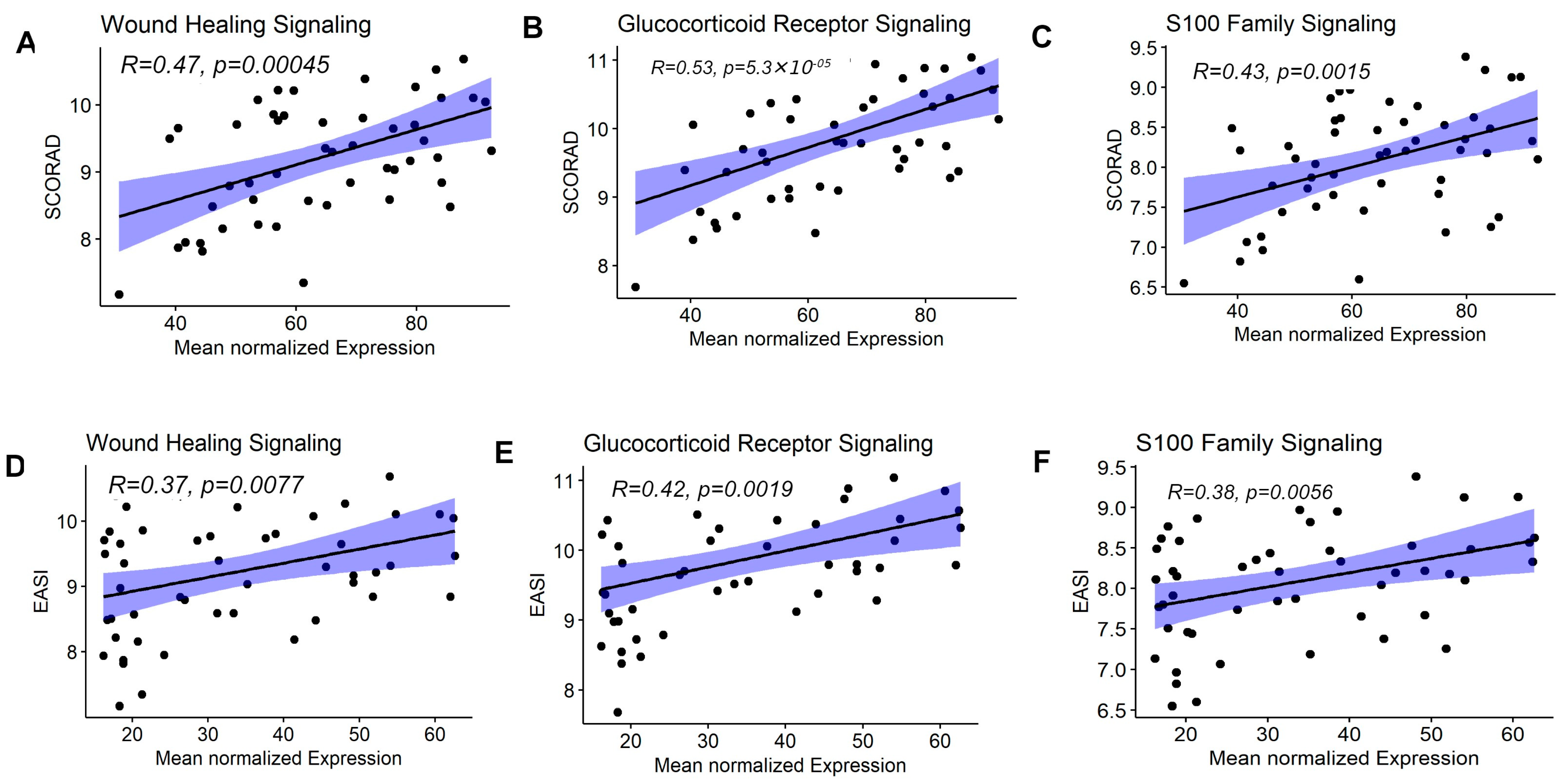
3.5. Enriched Pathway Signatures Were Correlated with AD Clinical Severity Measures
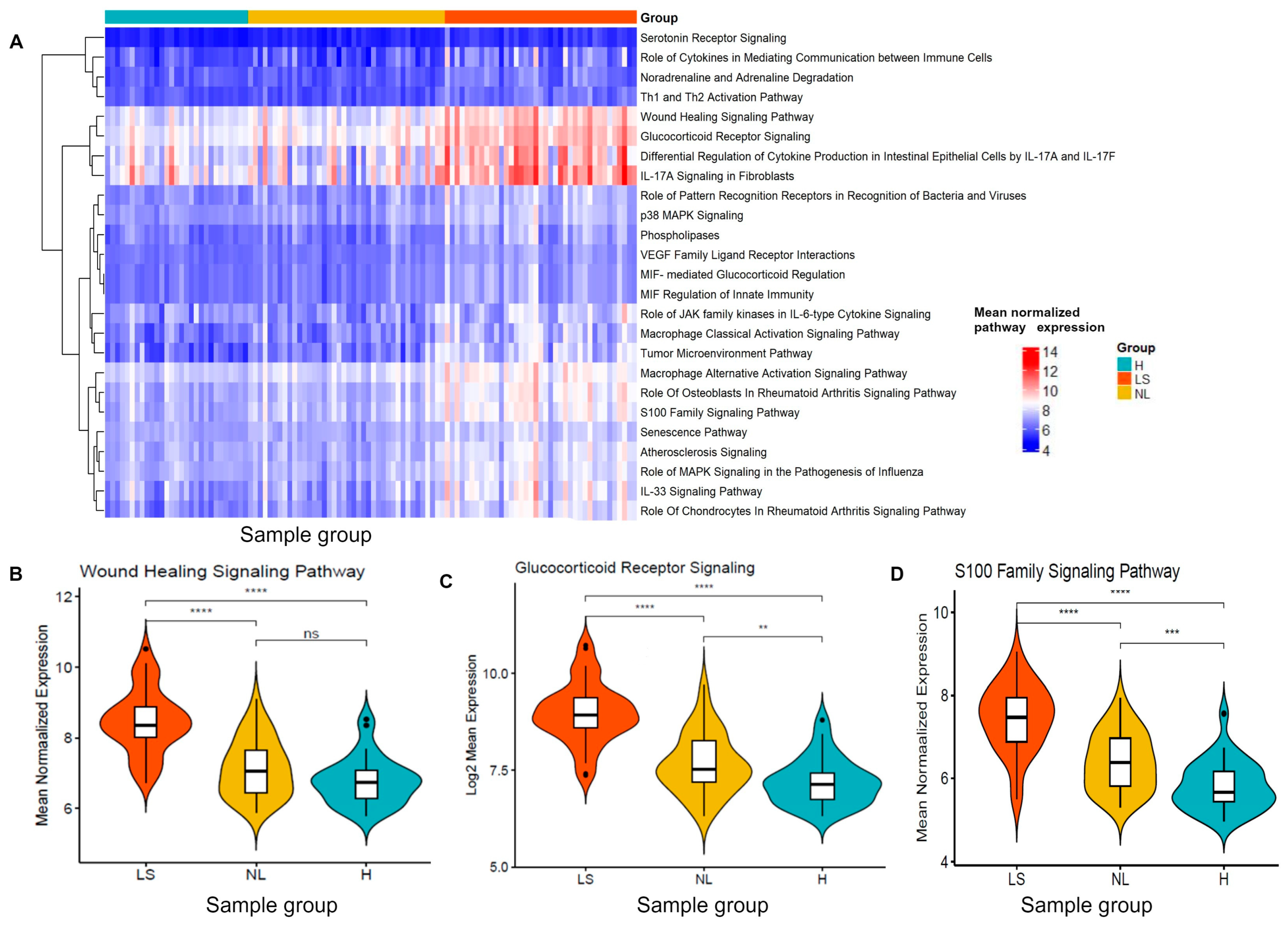
3.6. Validation of Pathways and Gene Signatures Discriminating Lesional, Non-Lesional AD and Healthy Skin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abuabara, K.; Yu, A.M.; Okhovat, J.P.; Allen, I.E.; Langan, S.M. The prevalence of atopic dermatitis beyond childhood: A systematic review and meta-analysis of longitudinal studies. Allergy 2018, 73, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Patel, K.R.; Singam, V.; Rastogi, S.; Silverberg, J.I. A systematic review and meta-analysis of the prevalence and phenotype of adult-onset atopic dermatitis. J. Am. Acad. Dermatol. 2019, 80, 1526–1532. [Google Scholar] [CrossRef] [PubMed]
- Aevermann, B.D.; Di Domizio, J.; Olah, P.; Saidoune, F.; Armstrong, J.M.; Bachelez, H.; Barker, J.; Haniffa, M.; Julia, V.; Juul, K.; et al. Cross-Comparison of Inflammatory Skin Disease Transcriptomics Identifies PTEN as a Pathogenic Disease Classifier in Cutaneous Lupus Erythematosus. J. Investig. Dermatol. 2024, 144, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Hamid, Q.; Boguniewicz, M.; Leung, D.Y. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J. Clin. Investig. 1994, 94, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.; Boguniewicz, M.; Howell, M.D.; Nomura, I.; Hamid, Q.A. New insights into atopic dermatitis. J. Clin. Investig. 2004, 113, 651–657. [Google Scholar] [CrossRef]
- Cork, M.J.; Danby, S.G.; Vasilopoulos, Y.; Hadgraft, J.; Lane, M.E.; Moustafa, M.; Guy, R.H.; MacGowan, A.L.; Tazi-Ahnini, R.; Ward, S.J. Epidermal barrier dysfunction in atopic dermatitis. J. Investig. Dermatol. 2009, 129, 1892–1908. [Google Scholar] [CrossRef]
- Janssens, M.; van Smeden, J.; Puppels, G.; Lavrijsen, A.; Caspers, P.; Bouwstra, J. Lipid to protein ratio plays an important role in the skin barrier function in patients with atopic eczema. Br. J. Dermatol. 2014, 170, 1248–1255. [Google Scholar] [CrossRef]
- Gruber, R.; Börnchen, C.; Rose, K.; Daubmann, A.; Volksdorf, T.; Wladykowski, E.; Vidal-Y-Sy, S.; Peters, E.M.; Danso, M.; Bouwstra, J.A.; et al. Diverse regulation of claudin-1 and claudin-4 in atopic dermatitis. Am. J. Pathol. 2015, 185, 2777–2789. [Google Scholar] [CrossRef]
- Dyjack, N.; Goleva, E.; Rios, C.; Kim, B.E.; Bin, L.; Taylor, P.; Bronchick, C.; Hall, C.F.; Richers, B.N.; Seibold, M.A.; et al. Minimally invasive skin tape strip RNA sequencing identifies novel characteristics of the type 2-high atopic dermatitis disease endotype. J. Allergy Clin. Immunol. 2018, 141, 1298–1309. [Google Scholar] [CrossRef]
- Martínez, B.A.; Shrotri, S.; Kingsmore, K.M.; Bachali, P.; Grammer, A.C.; Lipsky, P.E. Machine learning reveals distinct gene signature profiles in lesional and non-lesional regions of inflammatory skin diseases. Sci. Adv. 2022, 8, eabn4776. [Google Scholar] [CrossRef]
- Carrascosa-Carrillo, J.; Aterido, A.; Li, T.; Guillén, Y.; Martinez, S.; Marsal, S.; Julià, A. Toward Precision Medicine in Atopic Dermatitis Using Molecular-Based Approaches. Actas Dermo-Sifiliogr. 2024, 115, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Möbus, L.; Rodriguez, E.; Harder, I.; Boraczynski, N.; Szymczak, S.; Hübenthal, M.; Stölzl, D.; Gerdes, S.; Kleinheinz, A.; Abraham, S.; et al. Blood transcriptome profiling identifies 2 candidate endotypes of atopic dermatitis. J. Allergy Clin. Immunol. 2022, 150, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Acharjee, A.; Gribaleva, E.; Bano, S.; Gkoutos, G.V. Multi-omics-based identification of atopic dermatitis target genes and their potential associations with metabolites and miRNAs. Am. J. Transl. Res. 2021, 13, 13697–13709. [Google Scholar] [PubMed]
- Ewald, D.A.; Malajian, D.; Krueger, J.G.; Workman, C.T.; Wang, T.; Tian, S.; Litman, T.; Guttman-Yassky, E.; Suárez-Fariñas, M. Meta-analysis derived atopic dermatitis (MADAD) transcriptome defines a robust AD signature highlighting the involvement of atherosclerosis and lipid metabolism pathways. BMC Med. Genom. 2015, 8, 60. [Google Scholar] [CrossRef]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Fania, L.; Moretta, G.; Antonelli, F.; Scala, E.; Abeni, D.; Albanesi, C.; Madonna, S. Multiple Roles for Cytokines in Atopic Dermatitis: From Pathogenic Mediators to Endotype-Specific Biomarkers to Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 2684. [Google Scholar] [CrossRef]
- Silverberg, J.I.; Nelson, D.B.; Yosipovitch, G. Addressing treatment challenges in atopic dermatitis with novel topical therapies. J. Dermatol. Treat. 2016, 27, 568–576. [Google Scholar] [CrossRef]
- Zhong, Y.; Qin, K.; Li, L.; Liu, H.; Xie, Z.; Zeng, K. Identification of Immunological Biomarkers of Atopic Dermatitis by Integrated Analysis to Determine Molecular Targets for Diagnosis and Therapy. Int. J. Gen. Med. 2021, 14, 8193–8209. [Google Scholar] [CrossRef]
- Mankovich, N.; Kehoe, E.; Peterson, A.; Kirby, M. Pathway expression analysis. Sci Rep. 2022, 12, 21839. [Google Scholar] [CrossRef]
- Magnaye, K.M.; Clay, S.M.; Nicodemus-Johnson, J.; Naughton, K.A.; Huffman, J.; Altman, M.C.; Jackson, D.J.; Gern, J.E.; Hogarth, D.K.; Naureckas, E.T.; et al. DNA methylation signatures in airway cells from adult children of asthmatic mothers reflect subtypes of severe asthma. Proc. Natl. Acad. Sci. USA 2022, 119, e2116467119. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Rodriguez, E.; Degenhardt, F.; Baurecht, H.; Wehkamp, U.; Volks, N.; Szymczak, S.; Swindell, W.R.; Sarkar, M.K.; Raja, K.; et al. Atopic Dermatitis Is an IL-13–Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J. Investig. Dermatol. 2019, 139, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Rodriguez, E.; Stölzl, D.; Wehkamp, U.; Sun, J.; Gerdes, S.; Sarkar, M.K.; Hübenthal, M.; Zeng, C.; Uppala, R.; et al. Progression of acute-to-chronic atopic dermatitis is associated with quantitative rather than qualitative changes in cytokine responses. J. Allergy Clin. Immunol. 2020, 145, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Brunner, P.M.; Israel, A.; Zhang, N.; Leonard, A.; Wen, H.-C.; Huynh, T.; Tran, G.; Lyon, S.; Rodriguez, G.; Immaneni, S.; et al. Early-onset pediatric atopic dermatitis is characterized by TH2/TH17/TH22-centered inflammation and lipid alterations. J. Allergy Clin. Immunol. 2018, 141, 2094–2106. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Bissonnette, R.; Ungar, B.; Suárez-Fariñas, M.; Ardeleanu, M.; Esaki, H.; Suprun, M.; Estrada, Y.; Xu, H.; Peng, X.; et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.F.; Tycksen, E.D.; Sandell, L.J.; Brophy, R.H. Advantages of RNA-seq compared to RNA microarrays for transcriptome profiling of anterior cruciate ligament tears. J. Orthop. Res. 2018, 36, 484–497. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Chen, O. Identification of biomarkers and pathogenesis in severe asthma by coexpression network analysis. BMC Med. Genom. 2021, 14, 51. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Chen, L.; Qi, X.; Wang, J.; Yin, J.; Sun, P.; Sun, Y.; Wu, Y.; Zhang, L.; Gao, X. Identification of novel candidate genes and predicted miRNAs in atopic dermatitis patients by bioinformatic methods. Sci. Rep. 2022, 12, 22067. [Google Scholar] [CrossRef]
- Bang, H.; Kim, J.E.; Lee, H.S.; Park, S.M.; Park, D.-J.; Lee, E.J. Integrated bioinformatic analysis of gene expression profiling data to identify combinatorial biomarkers in inflammatory skin disease. Sci. Rep. 2022, 12, 5889. [Google Scholar] [CrossRef] [PubMed]
- Muzio, G.; O’Bray, L.; Borgwardt, K. Biological network analysis with deep learning. Brief. Bioinform. 2020, 22, 1515–1530. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Baizán, N.; Ribas, L.; Piferrer, F. Improved biomarker discovery through a plot twist in transcriptomic data analysis. BMC Biol. 2022, 20, 208. [Google Scholar] [CrossRef] [PubMed]
- Fuller, T.; Langfelder, P.; Presson, A.; Horvath, S. Review of Weighted Gene Coexpression Network Analysis. In Handbook of Statistical Bioinformatics; Lu, H.H.S., Schölkopf, B., Zhao, H., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 369–388. [Google Scholar]
- Fortino, V.; Wisgrill, L.; Werner, P.; Suomela, S.; Linder, N.; Jalonen, E.; Suomalainen, A.; Marwah, V.; Kero, M.; Pesonen, M.; et al. Machine-learning-driven biomarker discovery for the discrimination between allergic and irritant contact dermatitis. Proc. Natl. Acad. Sci. USA 2020, 117, 33474–33485. [Google Scholar] [CrossRef] [PubMed]
- Krizanova, O.; Penesova, A.; Sokol, J.; Hokynkova, A.; Samadian, A.; Babula, P. Signaling pathways in cutaneous wound healing. Front. Physiol. 2022, 13, 1030851. [Google Scholar] [CrossRef]
- Ungar, B.; Garcet, S.; Gonzalez, J.; Dhingra, N.; da Rosa, J.C.; Shemer, A.; Krueger, J.G.; Suarez-Farinas, M.; Guttman-Yassky, E. An Integrated Model of Atopic Dermatitis Biomarkers Highlights the Systemic Nature of the Disease. J. Investig. Dermatol. 2017, 137, 603–613. [Google Scholar] [CrossRef]
- Hamilton, J.D.; Suárez-Fariñas, M.; Dhingra, N.; Cardinale, I.; Li, X.; Kostic, A.; Ming, J.E.; Radin, A.R.; Krueger, J.G.; Graham, N.; et al. Dupilumab improves the molecular signature in skin of patients with moderate-to-severe atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 1293–1300. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Facheris, P.; Gomez-Arias, P.J.; Del Duca, E.; Da Rosa, J.C.; Weidinger, S.; Bissonnette, R.; Armstrong, A.W.; Seneschal, J.; Eyerich, K.; et al. Effect of abrocitinib on skin biomarkers in patients with moderate-to-severe atopic dermatitis. Allergy 2024, 79, 1258–1270. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Patrick, M.T.; Shuai, S.; Sarkar, M.K.; Chi, S.; Ruffino, B.; Billi, A.C.; Xing, X.; Uppala, R.; Zang, C.; et al. Cytokine responses in non-lesional psoriatic skin as clinical predictor to anti-TNF agents. J. Allergy Clin. Immunol. 2022, 149, 640–649.e645. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dessie, E.Y.; Ding, L.; Satish, L.; Mersha, T.B. Co-Expression Network and Machine Learning Analysis of Transcriptomics Data Identifies Distinct Gene Signatures and Pathways in Lesional and Non-Lesional Atopic Dermatitis. J. Pers. Med. 2024, 14, 960. https://doi.org/10.3390/jpm14090960
Dessie EY, Ding L, Satish L, Mersha TB. Co-Expression Network and Machine Learning Analysis of Transcriptomics Data Identifies Distinct Gene Signatures and Pathways in Lesional and Non-Lesional Atopic Dermatitis. Journal of Personalized Medicine. 2024; 14(9):960. https://doi.org/10.3390/jpm14090960
Chicago/Turabian StyleDessie, Eskezeia Y., Lili Ding, Latha Satish, and Tesfaye B. Mersha. 2024. "Co-Expression Network and Machine Learning Analysis of Transcriptomics Data Identifies Distinct Gene Signatures and Pathways in Lesional and Non-Lesional Atopic Dermatitis" Journal of Personalized Medicine 14, no. 9: 960. https://doi.org/10.3390/jpm14090960
APA StyleDessie, E. Y., Ding, L., Satish, L., & Mersha, T. B. (2024). Co-Expression Network and Machine Learning Analysis of Transcriptomics Data Identifies Distinct Gene Signatures and Pathways in Lesional and Non-Lesional Atopic Dermatitis. Journal of Personalized Medicine, 14(9), 960. https://doi.org/10.3390/jpm14090960