
Variation in CYP2A6 Activity and Personalized Medicine

Abstract
:1. CYP2A6 as a Pharmacogene
2. Variation in CYP2A6 Enzyme Activity
2.1. CYP2A6 Genetic Variation
2.2. Ethnic/Racial Differences in CYP2A6 Genetic Variation and CYP2A6 Activity
2.3. Factors That Regulate CYP2A6 Activity
2.3.1. Inducers
2.3.2. Inhibitors
2.4. Other Influences on CYP2A6
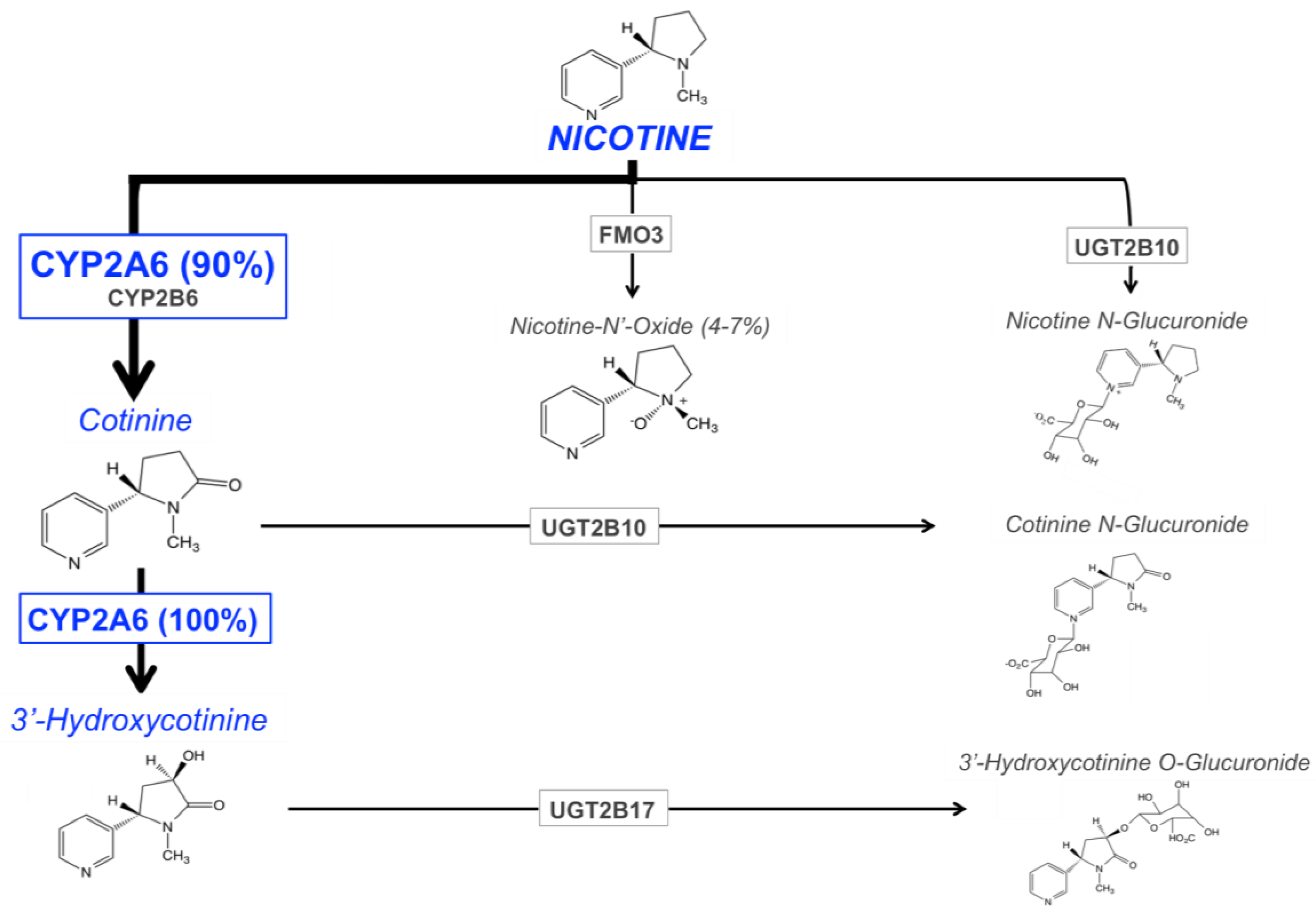
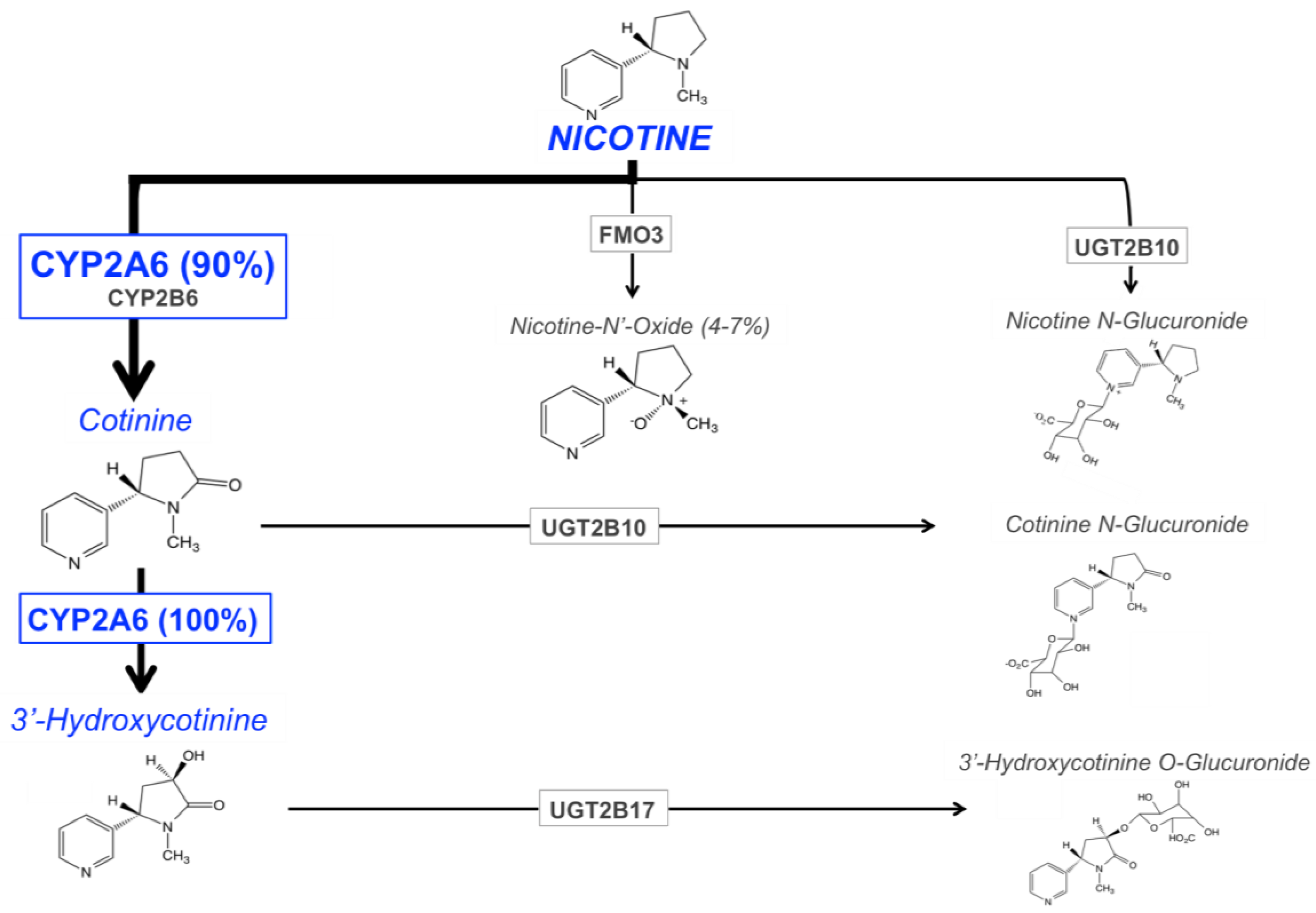
3. CYP2A6 and Nicotine Metabolism
3.1. Pathways of Nicotine Metabolism
3.2. Nicotine Metabolite Ratio
4. CYP2A6 and Smoking, Cessation, and Cancer
4.1. Smoking Behavior
4.2. Smoking Cessation
4.3. Lung Cancer Risk
5. CYP2A6 and Other Clinical Therapeutics
5.1. Tegafur
5.2. Letrozole
5.3. Efavirenz
5.4. Valproic Acid
5.5. Pilocarpine
5.6. Artemisinin and Artesunate
5.7. SM-12502
6. CYP2A6 and Dietary Substrates
6.1. Caffeine
6.2. Tyrosol
7. Conclusions and Areas of Future Research
Conflicts of Interest
References
- Koskela, S.; Hakkola, J.; Hukkanen, J.; Pelkonen, O.; Sorri, M.; Saranen, A.; Anttila, S.; Fernandez-Salguero, P.; Gonzalez, F.; Raunio, H. Expression of CYP2A genes in human liver and extrahepatic tissues. Biochem. Pharmacol. 1999, 57, 1407–1413. [Google Scholar] [CrossRef]
- McDonagh, E.M.; Wassenaar, C.; David, S.P.; Tyndale, R.F.; Altman, R.B.; Whirl-Carrillo, M.; Klein, T.E. PharmGKB summary: Very important pharmacogene information for cytochrome P-450, family 2, subfamily A, polypeptide 6. Pharmacogenet. Genom. 2012, 22, 695–708. [Google Scholar] [CrossRef]
- Loukola, A.; Buchwald, J.; Gupta, R.; Palviainen, T.; Hallfors, J.; Tikkanen, E.; Korhonen, T.; Ollikainen, M.; Sarin, A.P.; Ripatti, S.; et al. A Genome-Wide Association Study of a Biomarker of Nicotine Metabolism. PLoS Genet. 2015, 11, e1005498. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.A.; Prasad, B.; Claw, K.G.; Stapleton, P.; Chaudhry, A.; Schuetz, E.G.; Thummel, K.E.; Tyndale, R.F. Predictors of Variation in CYP2A6 mRNA, Protein, and Enzyme Activity in a Human Liver Bank: Influence of Genetic and Nongenetic Factors. J. Pharmacol. Exp. Ther. 2017, 360, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Hukkanen, J.; Jacob, P., III; Benowitz, N.L. Effect of grapefruit juice on cytochrome P450 2A6 and nicotine renal clearance. Clin. Pharmacol. Ther. 2006, 80, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Hakooz, N.; Hamdan, I. Effects of dietary broccoli on human in vivo caffeine metabolism: A pilot study on a group of Jordanian volunteers. Curr. Drug Metab. 2007, 8, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Higashi, E.; Fukami, T.; Itoh, M.; Kyo, S.; Inoue, M.; Yokoi, T.; Nakajima, M. Human CYP2A6 is induced by estrogen via estrogen receptor. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 1935–1941. [Google Scholar] [CrossRef] [PubMed]
- Koenigs, L.L.; Peter, R.M.; Thompson, S.J.; Rettie, A.E.; Trager, W.F. Mechanism-based inactivation of human liver cytochrome P450 2A6 by 8-methoxypsoralen. Drug Metab. Dispos. Biol. Fate Chem. 1997, 25, 1407–1415. [Google Scholar] [PubMed]
- Donato, M.T.; Viitala, P.; Rodriguez-Antona, C.; Lindfors, A.; Castell, J.V.; Raunio, H.; Gomez-Lechon, M.J.; Pelkonen, O. CYP2A5/CYP2A6 expression in mouse and human hepatocytes treated with various in vivo inducers. Drug Metab. Dispos. Biol. Fate Chem. 2000, 28, 1321–1326. [Google Scholar] [PubMed]
- Wong, H.L.; Murphy, S.E.; Hecht, S.S. Cytochrome P450 2A-catalyzed metabolic activation of structurally similar carcinogenic nitrosamines: N′-nitrosonornicotine enantiomers, N-nitrosopiperidine, and N-nitrosopyrrolidine. Chem. Res. Toxicol. 2005, 18, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Kushida, H.; Fujita, K.; Suzuki, A.; Yamada, M.; Endo, T.; Nohmi, T.; Kamataki, T. Metabolic activation of N-alkylnitrosamines in genetically engineered Salmonella typhimurium expressing CYP2E1 or CYP2A6 together with human NADPH-cytochrome P450 reductase. Carcinogenesis 2000, 21, 1227–1232. [Google Scholar] [PubMed]
- Messina, E.S.; Tyndale, R.F.; Sellers, E.M. A major role for CYP2A6 in nicotine C-oxidation by human liver microsomes. J. Pharmacol. Exp. Ther. 1997, 282, 1608–1614. [Google Scholar] [PubMed]
- Ikeda, K.; Yoshisue, K.; Matsushima, E.; Nagayama, S.; Kobayashi, K.; Tyson, C.A.; Chiba, K.; Kawaguchi, Y. Bioactivation of tegafur to 5-fluorouracil is catalyzed by cytochrome P-450 2A6 in human liver microsomes in vitro. Clin. Cancer Res. 2000, 6, 4409–4415. [Google Scholar] [PubMed]
- Murai, K.; Yamazaki, H.; Nakagawa, K.; Kawai, R.; Kamataki, T. Deactivation of anti-cancer drug letrozole to a carbinol metabolite by polymorphic cytochrome P450 2A6 in human liver microsomes. Xenobiotica 2009, 39, 795–802. [Google Scholar] [CrossRef] [PubMed]
- di Iulio, J.; Fayet, A.; Arab-Alameddine, M.; Rotger, M.; Lubomirov, R.; Cavassini, M.; Furrer, H.; Gunthard, H.F.; Colombo, S.; Csajka, C.; et al. In vivo analysis of efavirenz metabolism in individuals with impaired CYP2A6 function. Pharmacogenet. Genom. 2009, 19, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Aklillu, E.; McDonagh, E.M.; Klein, T.E.; Altman, R.B. PharmGKB summary: Caffeine pathway. Pharmacogenet. Genom. 2012, 22, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Yamamoto, T.; Nunoya, K.; Yokoi, T.; Nagashima, K.; Inoue, K.; Funae, Y.; Shimada, N.; Kamataki, T.; Kuroiwa, Y. Characterization of CYP2A6 involved in 3'-hydroxylation of cotinine in human liver microsomes. J. Pharmacol. Exp. Ther. 1996, 277, 1010–1015. [Google Scholar] [PubMed]
- Dempsey, D.; Tutka, P.; Jacob, P., III; Allen, F.; Schoedel, K.; Tyndale, R.F.; Benowitz, N.L. Nicotine metabolite ratio as an index of cytochrome P450 2A6 metabolic activity. Clin. Pharmacol. Ther. 2004, 76, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Swan, G.E.; Lessov-Schlaggar, C.N.; Bergen, A.W.; He, Y.; Tyndale, R.F.; Benowitz, N.L. Genetic and environmental influences on the ratio of 3′hydroxycotinine to cotinine in plasma and urine. Pharmacogenet. Genom. 2009, 19, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Baurley, J.W.; Edlund, C.K.; Pardamean, C.I.; Conti, D.V.; Krasnow, R.; Javitz, H.S.; Hops, H.; Swan, G.E.; Benowitz, N.L.; Bergen, A.W. Genome-wide association of the laboratory-based nicotine metabolite ratio in three ancestries. Nicotine Tob. Res. 2016, 18, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Hoffmann, E.; Zia, M.; Bodin, L.; Zeman, M.; Sellers, E.M.; Tyndale, R.F. Duplications and defects in the CYP2A6 gene: Identification, genotyping, and in vivo effects on smoking. Mol. Pharmacol. 2000, 58, 747–755. [Google Scholar] [PubMed]
- Fukami, T.; Nakajima, M.; Yamanaka, H.; Fukushima, Y.; McLeod, H.L.; Yokoi, T. A novel duplication type of CYP2A6 gene in African-American population. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Mwenifumbo, J.C.; Lessov-Schlaggar, C.N.; Zhou, Q.; Krasnow, R.E.; Swan, G.E.; Benowitz, N.L.; Tyndale, R.F. Identification of novel CYP2A6*1B variants: The CYP2A6*1B allele is associated with faster in vivo nicotine metabolism. Clin. Pharmacol. Ther. 2008, 83, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pitarque, M.; Ingelman-Sundberg, M. 3′-UTR polymorphism in the human CYP2A6 gene affects mRNA stability and enzyme expression. Biochem. Biophys. Res. Commun. 2006, 340, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Al Koudsi, N.; Hoffmann, E.B.; Assadzadeh, A.; Tyndale, R.F. Hepatic CYP2A6 levels and nicotine metabolism: Impact of genetic, physiological, environmental, and epigenetic factors. Eur. J. Clin. Pharmacol. 2010, 66, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Mwenifumbo, J.C.; Zhou, Q.; Benowitz, N.L.; Sellers, E.M.; Tyndale, R.F. New CYP2A6 gene deletion and conversion variants in a population of Black African descent. Pharmacogenomics 2010, 11, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Oscarson, M.; McLellan, R.A.; Gullsten, H.; Yue, Q.Y.; Lang, M.A.; Bernal, M.L.; Sinues, B.; Hirvonen, A.; Raunio, H.; Pelkonen, O.; et al. Characterisation and PCR-based detection of a CYP2A6 gene deletion found at a high frequency in a Chinese population. FEBS Lett. 1999, 448, 105–110. [Google Scholar] [CrossRef]
- Pitarque, M.; von Richter, O.; Oke, B.; Berkkan, H.; Oscarson, M.; Ingelman-Sundberg, M. Identification of a single nucleotide polymorphism in the TATA box of the CYP2A6 gene: Impairment of its promoter activity. Biochem. Biophys. Res. Commun. 2001, 284, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Oscarson, M.; McLellan, R.A.; Asp, V.; Ledesma, M.; Bernal Ruiz, M.L.; Sinues, B.; Rautio, A.; Ingelman-Sundberg, M. Characterization of a novel CYP2A7/CYP2A6 hybrid allele (CYP2A6*12) that causes reduced CYP2A6 activity. Hum. Mutat. 2002, 20, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Fukami, T.; Nakajima, M.; Higashi, E.; Yamanaka, H.; McLeod, H.L.; Yokoi, T. A novel CYP2A6*20 allele found in African-American population produces a truncated protein lacking enzymatic activity. Biochem. Pharmacol. 2005, 70, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Oscarson, M.; McLellan, R.A.; Gullsten, H.; Agundez, J.A.; Benitez, J.; Rautio, A.; Raunio, H.; Pelkonen, O.; Ingelman-Sundberg, M. Identification and characterisation of novel polymorphisms in the CYP2A locus: Implications for nicotine metabolism. FEBS Lett. 1999, 460, 321–327. [Google Scholar] [CrossRef]
- Xu, C.; Rao, Y.S.; Xu, B.; Hoffmann, E.; Jones, J.; Sellers, E.M.; Tyndale, R.F. An in vivo pilot study characterizing the new CYP2A6*7, *8, and *10 alleles. Biochem. Biophys. Res. Commun. 2002, 290, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Fukami, T.; Nakajima, M.; Higashi, E.; Yamanaka, H.; Sakai, H.; McLeod, H.L.; Yokoi, T. Characterization of novel CYP2A6 polymorphic alleles (CYP2A6*18 and CYP2A6*19) that affect enzymatic activity. Drug Metab. Dispos. Biol. Fate Chem. 2005, 33, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Mwenifumbo, J.C.; Al Koudsi, N.; Ho, M.K.; Zhou, Q.; Hoffmann, E.B.; Sellers, E.M.; Tyndale, R.F. Novel and established CYP2A6 alleles impair in vivo nicotine metabolism in a population of Black African descent. Hum. Mutat. 2008, 29, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.K.; Mwenifumbo, J.C.; Zhao, B.; Gillam, E.M.; Tyndale, R.F. A novel CYP2A6 allele, CYP2A6*23, impairs enzyme function in vitro and in vivo and decreases smoking in a population of Black-African descent. Pharmacogenet. Genom. 2008, 18, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Al Koudsi, N.; Ahluwalia, J.S.; Lin, S.K.; Sellers, E.M.; Tyndale, R.F. A novel CYP2A6 allele (CYP2A6*35) resulting in an amino-acid substitution (Asn438Tyr) is associated with lower CYP2A6 activity in vivo. Pharmacogenom. J. 2009, 9, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Hadidi, H.; Zahlsen, K.; Idle, J.R.; Cholerton, S. A single amino acid substitution (Leu160His) in cytochrome P450 CYP2A6 causes switching from 7-hydroxylation to 3-hydroxylation of coumarin. Food Chem. Toxicol. 1997, 35, 903–907. [Google Scholar] [CrossRef]
- Oscarson, M.; Gullsten, H.; Rautio, A.; Bernal, M.L.; Sinues, B.; Dahl, M.L.; Stengard, J.H.; Pelkonen, O.; Raunio, H.; Ingelman-Sundberg, M. Genotyping of human cytochrome P450 2A6 (CYP2A6), a nicotine C-oxidase. FEBS Lett. 1998, 438, 201–205. [Google Scholar] [CrossRef]
- Fukami, T.; Nakajima, M.; Yoshida, R.; Tsuchiya, Y.; Fujiki, Y.; Katoh, M.; McLeod, H.L.; Yokoi, T. A novel polymorphism of human CYP2A6 gene CYP2A6*17 has an amino acid substitution (V365M) that decreases enzymatic activity in vitro and in vivo. Clin. Pharmacol. Ther. 2004, 76, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Hosono, H.; Kumondai, M.; Maekawa, M.; Yamaguchi, H.; Mano, N.; Oda, A.; Hirasawa, N.; Hiratsuka, M. Functional Characterization of 34 CYP2A6 Allelic Variants by Assessment of Nicotine C-Oxidation and Coumarin 7-Hydroxylation Activities. Drug Metab. Dispos. Biol. Fate Chem. 2017, 45, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.A.; Chenoweth, M.J.; Tyndale, R.F. Pharmacogenetics of nicotine and associated smoking behaviors. Curr. Top. Behav. Neurosci. 2015, 23, 37–86. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.A.; Henderson, J.A.; Buchwald, D.; Howard, B.V.; Nez Henderson, P.; Tyndale, R.F. Variation in CYP2A6 and nicotine metabolism among two American Indian tribal groups differing in smoking patterns and risk for tobacco-related cancer. Pharmacogenet. Genom. 2017, 27, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.M.; Park, S.L.; Han, Y.; Wilkens, L.R.; Bickeboller, H.; Rosenberger, A.; Caporaso, N.; Landi, M.T.; Bruske, I.; Risch, A.; et al. Novel Association of Genetic Markers Affecting CYP2A6 Activity and Lung Cancer Risk. Cancer Res. 2016, 76, 5768–5776. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.A.; Zhu, A.Z.; Claw, K.G.; Prasad, B.; Korchina, V.; Hu, J.; Doddapaneni, H.V.; Muzny, D.; Schuetz, E.G.; Lerman, C.; et al. Novel CYP2A6 diplotypes identified through next-generation sequencing are associated with in-vitro and in-vivo nicotine metabolism. Pharmacogenet. Genom. 2017. In press. [Google Scholar]
- Chenoweth, M.J.; Ware, J.J.; Zhu, A.Z.X.; Cole, C.B.; Cox, L.S.; Nollen, N.; Ahluwalia, J.S.; Benowitz, N.L.; Schnoll, R.A.; Hawk, L.W., Jr.; et al. Genome-wide association study of a nicotine metabolism biomarker in African American smokers: Impact of chromosome 19 genetic influences. Addiction 2017. [Google Scholar] [CrossRef] [PubMed]
- Mwenifumbo, J.C.; Myers, M.G.; Wall, T.L.; Lin, S.K.; Sellers, E.M.; Tyndale, R.F. Ethnic variation in CYP2A6*7, CYP2A6*8 and CYP2A6*10 as assessed with a novel haplotyping method. Pharmacogenet. Genom. 2005, 15, 189–192. [Google Scholar] [CrossRef]
- Nakajima, M.; Fukami, T.; Yamanaka, H.; Higashi, E.; Sakai, H.; Yoshida, R.; Kwon, J.T.; McLeod, H.L.; Yokoi, T. Comprehensive evaluation of variability in nicotine metabolism and CYP2A6 polymorphic alleles in four ethnic populations. Clin. Pharmacol. Ther. 2006, 80, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.K.; Mwenifumbo, J.C.; Al Koudsi, N.; Okuyemi, K.S.; Ahluwalia, J.S.; Benowitz, N.L.; Tyndale, R.F. Association of nicotine metabolite ratio and CYP2A6 genotype with smoking cessation treatment in African-American light smokers. Clin. Pharmacol. Ther. 2009, 85, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Tiirikainen, M.I.; Patel, Y.M.; Wilkens, L.R.; Stram, D.O.; Le Marchand, L.; Murphy, S.E. Genetic determinants of CYP2A6 activity across racial/ethnic groups with different risks of lung cancer and effect on their smoking intensity. Carcinogenesis 2016, 37, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Schoedel, K.A.; Hoffmann, E.B.; Rao, Y.; Sellers, E.M.; Tyndale, R.F. Ethnic variation in CYP2A6 and association of genetically slow nicotine metabolism and smoking in adult Caucasians. Pharmacogenetics 2004, 14, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, N.L.; Perez-Stable, E.J.; Herrera, B.; Jacob, P., III. Slower metabolism and reduced intake of nicotine from cigarette smoking in Chinese-Americans. J. Natl. Cancer Inst. 2002, 94, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.L.; Shiffman, S.; Rait, M.A.; Benowitz, N.L. Race, gender, and nicotine metabolism in adolescent smokers. Nicotine Tob. Res. 2013, 15, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Perez-Stable, E.J.; Herrera, B.; Jacob, P., III; Benowitz, N.L. Nicotine metabolism and intake in black and white smokers. JAMA 1998, 280, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Ross, K.C.; Gubner, N.R.; Tyndale, R.F.; Hawk, L.W., Jr.; Lerman, C.; George, T.P.; Cinciripini, P.; Schnoll, R.A.; Benowitz, N.L. Racial differences in the relationship between rate of nicotine metabolism and nicotine intake from cigarette smoking. Pharmacol. Biochem. Behav. 2016, 148, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Binnington, M.J.; Zhu, A.Z.; Renner, C.C.; Lanier, A.P.; Hatsukami, D.K.; Benowitz, N.L.; Tyndale, R.F. CYP2A6 and CYP2B6 genetic variation and its association with nicotine metabolism in South Western Alaska Native people. Pharmacogenet. Genom. 2012, 22, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Rae, J.M.; Johnson, M.D.; Lippman, M.E.; Flockhart, D.A. Rifampin is a selective, pleiotropic inducer of drug metabolism genes in human hepatocytes: Studies with cDNA and oligonucleotide expression arrays. J. Pharmacol. Exp. Ther. 2001, 299, 849–857. [Google Scholar] [PubMed]
- Maurice, M.; Emiliani, S.; Dalet-Beluche, I.; Derancourt, J.; Lange, R. Isolation and characterization of a cytochrome P450 of the IIA subfamily from human liver microsomes. Eur. J. Biochem. 1991, 200, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Nakajima, M.; Higashi, E.; Yoshida, R.; Nagata, K.; Yamazoe, Y.; Yokoi, T. Induction of human CYP2A6 is mediated by the pregnane X receptor with peroxisome proliferator-activated receptor-γ coactivator 1α. J. Pharmacol. Exp. Ther. 2006, 319, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.B.; Parks, D.J.; Jones, S.A.; Bledsoe, R.K.; Consler, T.G.; Stimmel, J.B.; Goodwin, B.; Liddle, C.; Blanchard, S.G.; Willson, T.M.; et al. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J. Biol. Chem. 2000, 275, 15122–15127. [Google Scholar] [CrossRef] [PubMed]
- Onica, T.; Nichols, K.; Larin, M.; Ng, L.; Maslen, A.; Dvorak, Z.; Pascussi, J.M.; Vilarem, M.J.; Maurel, P.; Kirby, G.M. Dexamethasone-mediated up-regulation of human CYP2A6 involves the glucocorticoid receptor and increased binding of hepatic nuclear factor 4α to the proximal promoter. Mol. Pharmacol. 2008, 73, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, N.L.; Lessov-Schlaggar, C.N.; Swan, G.E.; Jacob, P., III. Female sex and oral contraceptive use accelerate nicotine metabolism. Clin. Pharmacol. Ther. 2006, 79, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, M.J.; Novalen, M.; Hawk, L.W., Jr.; Schnoll, R.A.; George, T.P.; Cinciripini, P.M.; Lerman, C.; Tyndale, R.F. Known and novel sources of variability in the nicotine metabolite ratio in a large sample of treatment-seeking smokers. Cancer Epidemiol. Biomark. Prev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Siu, E.C.; Tyndale, R.F. Selegiline is a mechanism-based inactivator of CYP2A6 inhibiting nicotine metabolism in humans and mice. J. Pharmacol. Exp. Ther. 2008, 324, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Tzaneva, S.; Kittler, H.; Thallinger, C.; Honigsmann, H.; Tanew, A. Oral vs. bath PUVA using 8-methoxypsoralen for chronic palmoplantar eczema. Photodermatol. Photoimmunol. Photomed. 2009, 25, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.B.; McCarty, J.R.; Jarratt, M. Treatment of psoriasis with 8-methoxypsoralen and sunlight. South Med. J. 1978, 71, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Cereda, E.; Cilia, R.; Canesi, M.; Tesei, S.; Mariani, C.B.; Zecchinelli, A.L.; Pezzoli, G. Efficacy of rasagiline and selegiline in Parkinson’s disease: A head-to-head 3-year retrospective case-control study. J. Neurol. 2017, 264, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Blobaum, A.L. Mechanism-based inactivation and reversibility: Is there a new trend in the inactivation of cytochrome p450 enzymes? Drug Metab. Dispos. 2006, 34, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Kilicarslan, T.; Tyndale, R.F.; Sellers, E.M. Evaluation of methoxsalen, tranylcypromine, and tryptamine as specific and selective CYP2A6 inhibitors in vitro. Drug Metab. Dispos. Biol. Fate Chem. 2001, 29, 897–902. [Google Scholar] [PubMed]
- Draper, A.J.; Madan, A.; Parkinson, A. Inhibition of coumarin 7-hydroxylase activity in human liver microsomes. Arch. Biochem. Biophys. 1997, 341, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ramamoorthy, Y.; Kilicarslan, T.; Nolte, H.; Tyndale, R.F.; Sellers, E.M. Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab. Dispos. Biol. Fate Chem. 2002, 30, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.P.; McDonnell, L.; Hardiman, O.M.; Farrell, M.A.; Phillips, J.P.; Tipton, K.F. The oxidation of tryptamine by the two forms of monoamine oxidase in human tissues. Biochem. Pharmacol. 1986, 35, 3255–3260. [Google Scholar] [CrossRef]
- Higashi, E.; Nakajima, M.; Katoh, M.; Tokudome, S.; Yokoi, T. Inhibitory effects of neurotransmitters and steroids on human CYP2A6. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 508–514. [Google Scholar] [CrossRef] [PubMed]
- MacDougall, J.M.; Fandrick, K.; Zhang, X.; Serafin, S.V.; Cashman, J.R. Inhibition of human liver microsomal (S)-nicotine oxidation by (-)-menthol and analogues. Chem. Res. Toxicol. 2003, 16, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, N.L.; Herrera, B.; Jacob, P., III. Mentholated cigarette smoking inhibits nicotine metabolism. J. Pharmacol. Exp. Ther. 2004, 310, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.J.; Tanner, J.A.; Taylor, A.E.; Bin, Z.; Haycock, P.; Bowden, J.; Rogers, P.J.; Davey Smith, G.; Tyndale, R.F.; Munafo, M.R. Does coffee consumption impact on heaviness of smoking? Addiction 2017. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Oshiro, T.; Thomas, S.; Higa, A.; Black, S.; Todorovic, A.; Elbarbry, F.; Harrelson, J.P. Inactivation of CYP2A6 by the Dietary Phenylpropanoid trans-Cinnamic Aldehyde (Cinnamaldehyde) and Estimation of Interactions with Nicotine and Letrozole. Drug Metab. Dispos. Biol. Fate Chem. 2016, 44, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Molander, L.; Hansson, A.; Lunell, E. Pharmacokinetics of nicotine in healthy elderly people. Clin. Pharmacol. Ther. 2001, 69, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Gourlay, S.G.; Benowitz, N.L.; Forbes, A.; McNeil, J.J. Determinants of plasma concentrations of nicotine and cotinine during cigarette smoking and transdermal nicotine treatment. Eur. J. Clin. Pharmacol. 1997, 51, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zhuo, W.; He, Y.J.; Zhou, H.H.; Fan, L. Pharmacogenetics of P450 oxidoreductase: Implications in drug metabolism and therapy. Pharmacogenet. Genom. 2012, 22, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Weng, Y.; Zhang, Q.Y.; Cui, H.; Behr, M.; Wu, L.; Yang, W.; Zhang, L.; Ding, X. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: Impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J. Biol. Chem. 2003, 278, 25895–25901. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.J.; Otto, D.M.; Carrie, D.; Magnuson, M.A.; McLaren, A.W.; Rosewell, I.; Wolf, C.R. Inactivation of the hepatic cytochrome P450 system by conditional deletion of hepatic cytochrome P450 reductase. J. Biol. Chem. 2003, 278, 13480–13486. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, M.J.; Zhu, A.Z.; Sanderson Cox, L.; Ahluwalia, J.S.; Benowitz, N.L.; Tyndale, R.F. Variation in P450 oxidoreductase (POR) A503V and flavin-containing monooxygenase (FMO)-3 E158K is associated with minor alterations in nicotine metabolism, but does not alter cigarette consumption. Pharmacogenet. Genom. 2014, 24, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, E.G.; Strom, S.; Yasuda, K.; Lecureur, V.; Assem, M.; Brimer, C.; Lamba, J.; Kim, R.B.; Ramachandran, V.; Komoroski, B.J.; et al. Disrupted bile acid homeostasis reveals an unexpected interaction among nuclear hormone receptors, transporters, and cytochrome P450. J. Biol. Chem. 2001, 276, 39411–39418. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Lukacik, P.; Guo, K.; Ugochukwu, E.; Kavanagh, K.L.; Marsden, B.; Oppermann, U. Structure-activity relationships of human AKR-type oxidoreductases involved in bile acid synthesis: AKR1D1 and AKR1C4. Mol. Cell. Endocrinol. 2009, 301, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Rizner, T.L.; Penning, T.M. Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids 2014, 79, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, B.; Molony, C.; Chudin, E.; Hao, K.; Zhu, J.; Gaedigk, A.; Suver, C.; Zhong, H.; Leeder, J.S.; et al. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010, 20, 1020–1036. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.S.; Thirumaran, R.K.; Yasuda, K.; Yang, X.; Fan, Y.; Strom, S.C.; Schuetz, E.G. Genetic variation in aldo-keto reductase 1D1 (AKR1D1) affects the expression and activity of multiple cytochrome P450s. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 1538–1547. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, N.L.; Jacob, P., III. Metabolism of nicotine to cotinine studied by a dual stable isotope method. Clin. Pharmacol. Ther. 1994, 56, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, N.L.; Jacob, P., III; Fong, I.; Gupta, S. Nicotine metabolic profile in man: Comparison of cigarette smoking and transdermal nicotine. J. Pharmacol. Exp. Ther. 1994, 268, 296–303. [Google Scholar] [PubMed]
- Byrd, G.D.; Chang, K.M.; Greene, J.M.; deBethizy, J.D. Evidence for urinary excretion of glucuronide conjugates of nicotine, cotinine, and trans-3′-hydroxycotinine in smokers. Drug Metab. Dispos. Biol. Fate Chem. 1992, 20, 192–197. [Google Scholar] [PubMed]
- Brandange, S.; Lindblom, L. The enzyme “aldehyde oxidase“ is an iminium oxidase. Reaction with nicotine ∆ 1′(5′) iminium ion. Biochem. Biophys. Res. Commun. 1979, 91, 991–996. [Google Scholar] [CrossRef]
- Benowitz, N.L.; Jacob, P., III. Trans-3′-hydroxycotinine: Disposition kinetics, effects and plasma levels during cigarette smoking. Br. J. Clin. Pharmacol. 2001, 51, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, N.L.; Jacob, P., III. Effects of cigarette smoking and carbon monoxide on nicotine and cotinine metabolism. Clin. Pharmacol. Ther. 2000, 67, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Zevin, S.; Jacob, P., III; Benowitz, N. Cotinine effects on nicotine metabolism. Clin. Pharmacol. Ther. 1997, 61, 649–654. [Google Scholar] [CrossRef]
- Al Koudsi, N.; Tyndale, R.F. Hepatic CYP2B6 is altered by genetic, physiologic, and environmental factors but plays little role in nicotine metabolism. Xenobiotica 2010, 40, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Inoue, K.; Hashimoto, M.; Shimada, T. Roles of CYP2A6 and CYP2B6 in nicotine C-oxidation by human liver microsomes. Arch. Toxicol. 1999, 73, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Kaivosaari, S.; Toivonen, P.; Hesse, L.M.; Koskinen, M.; Court, M.H.; Finel, M. Nicotine glucuronidation and the human UDP-glucuronosyltransferase UGT2B10. Mol. Pharmacol. 2007, 72, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Tanaka, E.; Kwon, J.T.; Yokoi, T. Characterization of nicotine and cotinine N-glucuronidations in human liver microsomes. Drug Metab. Dispos. Biol. Fate Chem. 2002, 30, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, G.E.; Murphy, S.E. N-glucuronidation of nicotine and cotinine by human liver microsomes and heterologously expressed UDP-glucuronosyltransferases. Drug Metab. Dispos. Biol. Fate Chem. 2003, 31, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Izukawa, T.; Nakajima, M.; Fujiwara, R.; Yamanaka, H.; Fukami, T.; Takamiya, M.; Aoki, Y.; Ikushiro, S.; Sakaki, T.; Yokoi, T. Quantitative analysis of UDP-glucuronosyltransferase (UGT) 1A and UGT2B expression levels in human livers. Drug Metab. Dispos. Biol. Fate Chem. 2009, 37, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Hukkanen, J.; Jacob, P., III; Benowitz, N.L. Metabolism and disposition kinetics of nicotine. Pharmacol. Rev. 2005, 57, 79–115. [Google Scholar] [CrossRef] [PubMed]
- Mooney, M.E.; Li, Z.Z.; Murphy, S.E.; Pentel, P.R.; Le, C.; Hatsukami, D.K. Stability of the nicotine metabolite ratio in ad libitum and reducing smokers. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1396–1400. [Google Scholar] [CrossRef] [PubMed]
- St Helen, G.; Jacob, P., III; Benowitz, N.L. Stability of the nicotine metabolite ratio in smokers of progressively reduced nicotine content cigarettes. Nicotine Tob. Res. 2013, 15, 1939–1942. [Google Scholar] [CrossRef] [PubMed]
- Lea, R.A.; Dickson, S.; Benowitz, N.L. Within-subject variation of the salivary 3HC/COT ratio in regular daily smokers: Prospects for estimating CYP2A6 enzyme activity in large-scale surveys of nicotine metabolic rate. J. Anal. Toxicol. 2006, 30, 386–389. [Google Scholar] [CrossRef] [PubMed]
- St Helen, G.; Novalen, M.; Heitjan, D.F.; Dempsey, D.; Jacob, P., III; Aziziyeh, A.; Wing, V.C.; George, T.P.; Tyndale, R.F.; Benowitz, N.L. Reproducibility of the nicotine metabolite ratio in cigarette smokers. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.A.; Novalen, M.; Jatlow, P.; Huestis, M.A.; Murphy, S.E.; Kaprio, J.; Kankaanpaa, A.; Galanti, L.; Stefan, C.; George, T.P.; et al. Nicotine metabolite ratio (3-hydroxycotinine/cotinine) in plasma and urine by different analytical methods and laboratories: Implications for clinical implementation. Cancer Epidemiol. Biomark. Prev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Nakajima-Taniguchi, C.; Fukuda, T.; Funamoto, M.; Maeda, M.; Tange, E.; Ueki, R.; Kawashima, K.; Hara, H.; Fujio, Y.; et al. CYP2A6 polymorphisms are associated with nicotine dependence and influence withdrawal symptoms in smoking cessation. Pharmacogenomics J. 2006, 6, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, C.A.; Dong, Q.; Wei, Q.; Amos, C.I.; Spitz, M.R.; Tyndale, R.F. Relationship between CYP2A6 and CHRNA5-CHRNA3-CHRNB4 variation and smoking behaviors and lung cancer risk. J. Natl. Cancer Inst. 2011, 103, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Sofuoglu, M.; Herman, A.I.; Nadim, H.; Jatlow, P. Rapid nicotine clearance is associated with greater reward and heart rate increases from intravenous nicotine. Neuropsychopharmacology 2012, 37, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, E.; Benowitz, N.; Cargill, A.; Jacob, R.; Hinks, L.; Day, I.; Murphy, M.; Walton, R. Determinants of the rate of nicotine metabolism and effects on smoking behavior. Clin. Pharmacol. Ther. 2006, 80, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Schnoll, R.A.; Patterson, F.; Wileyto, E.P.; Tyndale, R.F.; Benowitz, N.; Lerman, C. Nicotine metabolic rate predicts successful smoking cessation with transdermal nicotine: A validation study. Pharmacol. Biochem. Behav. 2009, 92, 6–11. [Google Scholar] [CrossRef] [PubMed]
- McMorrow, M.J.; Foxx, R.M. Nicotine’s role in smoking: An analysis of nicotine regulation. Psychol. Bull. 1983, 93, 302–327. [Google Scholar] [CrossRef] [PubMed]
- Jarvik, M.E.; Madsen, D.C.; Olmstead, R.E.; Iwamoto-Schaap, P.N.; Elins, J.L.; Benowitz, N.L. Nicotine blood levels and subjective craving for cigarettes. Pharmacol. Biochem. Behav. 2000, 66, 553–558. [Google Scholar] [CrossRef]
- Ariyoshi, N.; Miyamoto, M.; Umetsu, Y.; Kunitoh, H.; Dosaka-Akita, H.; Sawamura, Y.; Yokota, J.; Nemoto, N.; Sato, K.; Kamataki, T. Genetic polymorphism of CYP2A6 gene and tobacco-induced lung cancer risk in male smokers. Cancer Epidemiol. Biomark. Prev. 2002, 11, 890–894. [Google Scholar]
- Fujieda, M.; Yamazaki, H.; Saito, T.; Kiyotani, K.; Gyamfi, M.A.; Sakurai, M.; Dosaka-Akita, H.; Sawamura, Y.; Yokota, J.; Kunitoh, H.; et al. Evaluation of CYP2A6 genetic polymorphisms as determinants of smoking behavior and tobacco-related lung cancer risk in male Japanese smokers. Carcinogenesis 2004, 25, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Yang, X.; Li, S.; Jia, C. Association of CYP2A6 gene polymorphisms with cigarette consumption: A meta-analysis. Drug Alcohol Depend. 2015, 149, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, N.L.; Pomerleau, O.F.; Pomerleau, C.S.; Jacob, P., III. Nicotine metabolite ratio as a predictor of cigarette consumption. Nicotine Tob. Res. 2003, 5, 621–624. [Google Scholar] [PubMed]
- Strasser, A.A.; Benowitz, N.L.; Pinto, A.G.; Tang, K.Z.; Hecht, S.S.; Carmella, S.G.; Tyndale, R.F.; Lerman, C.E. Nicotine metabolite ratio predicts smoking topography and carcinogen biomarker level. Cancer Epidemiol. Biomark. Prev. 2011, 20, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.A.; Malaiyandi, V.; Hoffmann, E.; Tyndale, R.F.; Lerman, C. An association of CYP2A6 genotype and smoking topography. Nicotine Tob. Res. 2007, 9, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.K.; Faseru, B.; Choi, W.S.; Nollen, N.L.; Mayo, M.S.; Thomas, J.L.; Okuyemi, K.S.; Ahluwalia, J.S.; Benowitz, N.L.; Tyndale, R.F. Utility and relationships of biomarkers of smoking in African-American light smokers. Cancer Epidemiol. Biomark. Prev. 2009, 18, 3426–3434. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.Z.; Binnington, M.J.; Renner, C.C.; Lanier, A.P.; Hatsukami, D.K.; Stepanov, I.; Watson, C.H.; Sosnoff, C.S.; Benowitz, N.L.; Tyndale, R.F. Alaska Native smokers and smokeless tobacco users with slower CYP2A6 activity have lower tobacco consumption, lower tobacco-specific nitrosamine exposure and lower tobacco-specific nitrosamine bioactivation. Carcinogenesis 2013, 34, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.F.; Hinks, L.J.; Morton, N.E.; Day, I.N. The use of long PCR to confirm three common alleles at the CYP2A6 locus and the relationship between genotype and smoking habit. Ann. Hum. Genet. 2000, 64, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, M.J.; O’Loughlin, J.; Sylvestre, M.P.; Tyndale, R.F. CYP2A6 slow nicotine metabolism is associated with increased quitting by adolescent smokers. Pharmacogenet. Genom. 2013, 23, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Lerman, C.; Tyndale, R.; Patterson, F.; Wileyto, E.P.; Shields, P.G.; Pinto, A.; Benowitz, N. Nicotine metabolite ratio predicts efficacy of transdermal nicotine for smoking cessation. Clin. Pharmacol. Ther. 2006, 79, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Warner, C.; Shoaib, M. How does bupropion work as a smoking cessation aid? Addict. Biol. 2005, 10, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.S.; Bloom, A.J.; Baker, T.B.; Smith, S.S.; Piper, M.E.; Martinez, M.; Saccone, N.; Hatsukami, D.; Goate, A.; Bierut, L. Pharmacotherapy effects on smoking cessation vary with nicotine metabolism gene (CYP2A6). Addiction 2014, 109, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Patterson, F.; Schnoll, R.A.; Wileyto, E.P.; Pinto, A.; Epstein, L.H.; Shields, P.G.; Hawk, L.W.; Tyndale, R.F.; Benowitz, N.; Lerman, C. Toward personalized therapy for smoking cessation: A randomized placebo-controlled trial of bupropion. Clin. Pharmacol. Ther. 2008, 84, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Lerman, C.; Schnoll, R.A.; Hawk, L.W., Jr.; Cinciripini, P.; George, T.P.; Wileyto, E.P.; Swan, G.E.; Benowitz, N.L.; Heitjan, D.F.; Tyndale, R.F.; et al. Use of the nicotine metabolite ratio as a genetically informed biomarker of response to nicotine patch or varenicline for smoking cessation: A randomised, double-blind placebo-controlled trial. Lancet Respir. Med. 2015, 3, 131–138. [Google Scholar] [CrossRef]
- Garrison, G.D.; Dugan, S.E. Varenicline: A first-line treatment option for smoking cessation. Clin. Ther. 2009, 31, 463–491. [Google Scholar] [CrossRef] [PubMed]
- Khuder, S.A. Effect of cigarette smoking on major histological types of lung cancer: A meta-analysis. Lung Cancer 2001, 31, 139–148. [Google Scholar] [CrossRef]
- Hecht, S.S. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998, 11, 559–603. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, C.A.; Ye, Y.; Cai, Q.; Aldrich, M.C.; Knight, J.; Spitz, M.R.; Wu, X.; Blot, W.J.; Tyndale, R.F. CYP2A6 reduced activity gene variants confer reduction in lung cancer risk in African American smokers—findings from two independent populations. Carcinogenesis 2015, 36, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Murphy, S.E.; Wilkens, L.R.; Stram, D.O.; Hecht, S.S.; Le Marchand, L. Association of CYP2A6 activity with lung cancer incidence in smokers: The multiethnic cohort study. PLoS ONE 2017, 12, e0178435. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Yamazaki, H.; Shimada, N.; Nakajima, M.; Yokoi, T. Roles of cytochromes P450 1A2, 2A6, and 2C8 in 5-fluorouracil formation from tegafur, an anticancer prodrug, in human liver microsomes. Drug Metab. Dispos. Biol. Fate Chem. 2000, 28, 1457–1463. [Google Scholar] [PubMed]
- Wang, H.; Bian, T.; Liu, D.; Jin, T.; Chen, Y.; Lin, A.; Chen, C. Association analysis of CYP2A6 genotypes and haplotypes with 5-fluorouracil formation from tegafur in human liver microsomes. Pharmacogenomics 2011, 12, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Kaida, Y.; Inui, N.; Suda, T.; Nakamura, H.; Watanabe, H.; Chida, K. The CYP2A6*4 allele is determinant of S-1 pharmacokinetics in Japanese patients with non-small-cell lung cancer. Clin. Pharmacol. Ther. 2008, 83, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Fujita, K.; Nishimura, K.; Ishida, H.; Yamashita, K.; Sunakawa, Y.; Mizuno, K.; Miwa, K.; Nagashima, F.; Tanigawara, Y.; et al. Pharmacokinetics of S-1 and CYP2A6 genotype in Japanese patients with advanced cancer. Oncol. Rep. 2010, 24, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Yamamoto, W.; Endo, S.; Endo, H.; Nagashima, F.; Ichikawa, W.; Tanaka, R.; Miya, T.; Araki, K.; Kodama, K.; et al. CYP2A6 and the plasma level of 5-chloro-2, 4-dihydroxypyridine are determinants of the pharmacokinetic variability of tegafur and 5-fluorouracil, respectively, in Japanese patients with cancer given S-1. Cancer Sci. 2008, 99, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.Y.; Lim, H.S.; Nam, B.H.; Kook, M.C.; Kim, Y.W.; Ryu, K.W.; Lee, J.H.; Choi, I.J.; Lee, J.S.; Park, Y.I.; et al. Association of CYP2A6 polymorphisms with S-1 plus docetaxel therapy outcomes in metastatic gastric cancer. Pharmacogenomics 2009, 10, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Park, S.R.; Ahn, Y.; Ryu, M.H.; Ryoo, B.Y.; Kong, S.Y.; Yook, J.H.; Yoo, M.W.; Kim, B.S.; Kim, B.S.; et al. Associations between CYP2A6 polymorphisms and outcomes of adjuvant S-1 chemotherapy in patients with curatively resected gastric cancer. Gastric Cancer 2017, 20, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Hong, Y.S.; Shim, E.K.; Kong, S.Y.; Shin, A.; Baek, J.Y.; Jung, K.H. S-1 plus irinotecan and oxaliplatin for the first-line treatment of patients with metastatic colorectal cancer: A prospective phase II study and pharmacogenetic analysis. Br. J. Cancer 2013, 109, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.P.; Jang, G.; Hong, Y.S.; Lim, H.S.; Bae, K.S.; Kim, H.S.; Lee, S.S.; Shin, J.G.; Lee, J.L.; Ryu, M.H.; et al. Phase II study of S-1 combined with oxaliplatin as therapy for patients with metastatic biliary tract cancer: Influence of the CYP2A6 polymorphism on pharmacokinetics and clinical activity. Br. J. Cancer 2011, 104, 605–612. [Google Scholar] [CrossRef] [PubMed]
- He, M.M.; Zhang, D.S.; Wang, F.; Wang, Z.X.; Yuan, S.Q.; Wang, Z.Q.; Luo, H.Y.; Ren, C.; Qiu, M.Z.; Jin, Y.; et al. Phase II trial of S-1 plus leucovorin in patients with advanced gastric cancer and clinical prediction by S-1 pharmacogenetic pathway. Cancer Chemother. Pharmacol. 2017, 79, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Baek, J.Y.; Oh, J.H.; Park, S.C.; Sohn, D.K.; Kim, M.J.; Chang, H.J.; Kong, S.Y.; Kim, D.Y. A phase II study of preoperative chemoradiation with tegafur-uracil plus leucovorin for locally advanced rectal cancer with pharmacogenetic analysis. Radiat. Oncol. 2017, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, A.S. The discovery and mechanism of action of letrozole. Breast Cancer Res. Treat. 2007, 105 (Suppl. 1), 7–17. [Google Scholar] [CrossRef] [PubMed]
- Desta, Z.; Kreutz, Y.; Nguyen, A.T.; Li, L.; Skaar, T.; Kamdem, L.K.; Henry, N.L.; Hayes, D.F.; Storniolo, A.M.; Stearns, V.; et al. Plasma letrozole concentrations in postmenopausal women with breast cancer are associated with CYP2A6 genetic variants, body mass index, and age. Clin. Pharmacol. Ther. 2011, 90, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Tanii, H.; Shitara, Y.; Horie, T. Population pharmacokinetic analysis of letrozole in Japanese postmenopausal women. Eur. J. Clin. Pharmacol. 2011, 67, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Marzolini, C.; Telenti, A.; Decosterd, L.A.; Greub, G.; Biollaz, J.; Buclin, T. Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS 2001, 15, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Ogburn, E.T.; Jones, D.R.; Masters, A.R.; Xu, C.; Guo, Y.; Desta, Z. Efavirenz primary and secondary metabolism in vitro and in vivo: Identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 1218–1229. [Google Scholar] [CrossRef] [PubMed]
- Heil, S.G.; van der Ende, M.E.; Schenk, P.W.; van der Heiden, I.; Lindemans, J.; Burger, D.; van Schaik, R.H. Associations between ABCB1, CYP2A6, CYP2B6, CYP2D6, and CYP3A5 alleles in relation to efavirenz and nevirapine pharmacokinetics in HIV-infected individuals. Ther. Drug Monit. 2012, 34, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Sukasem, C.; Chamnanphon, M.; Koomdee, N.; Santon, S.; Jantararoungtong, T.; Prommas, S.; Puangpetch, A.; Manosuthi, W. Pharmacogenetics and clinical biomarkers for subtherapeutic plasma efavirenz concentration in HIV-1 infected Thai adults. Drug Metab. Pharmacokinet. 2014, 29, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Sinxadi, P.Z.; Leger, P.D.; McIlleron, H.M.; Smith, P.J.; Dave, J.A.; Levitt, N.S.; Maartens, G.; Haas, D.W. Pharmacogenetics of plasma efavirenz exposure in HIV-infected adults and children in South Africa. Br. J. Clin. Pharmacol. 2015, 80, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Cusato, J.; Tomasello, C.; Simiele, M.; Calcagno, A.; Bonora, S.; Marinaro, L.; Leggieri, A.; Allegra, S.; Di Perri, G.; D’Avolio, A. Efavirenz pharmacogenetics in a cohort of Italian patients. Int. J. Antimicrob. Agents 2016, 47, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Swart, M.; Evans, J.; Skelton, M.; Castel, S.; Wiesner, L.; Smith, P.J.; Dandara, C. An Expanded Analysis of Pharmacogenetics Determinants of Efavirenz Response that Includes 3′-UTR Single Nucleotide Polymorphisms among Black South African HIV/AIDS Patients. Front. Genet. 2015, 6, 356. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.W.; Kwara, A.; Richardson, D.M.; Baker, P.; Papageorgiou, I.; Acosta, E.P.; Morse, G.D.; Court, M.H. Secondary metabolism pathway polymorphisms and plasma efavirenz concentrations in HIV-infected adults with CYP2B6 slow metabolizer genotypes. J. Antimicrob. Chemother. 2014, 69, 2175–2182. [Google Scholar] [CrossRef] [PubMed]
- Kwara, A.; Lartey, M.; Sagoe, K.W.; Kenu, E.; Court, M.H. CYP2B6, CYP2A6 and UGT2B7 genetic polymorphisms are predictors of efavirenz mid-dose concentration in HIV-infected patients. AIDS 2009, 23, 2101–2106. [Google Scholar] [CrossRef] [PubMed]
- Sarfo, F.S.; Zhang, Y.; Egan, D.; Tetteh, L.A.; Phillips, R.; Bedu-Addo, G.; Sarfo, M.A.; Khoo, S.; Owen, A.; Chadwick, D.R. Pharmacogenetic associations with plasma efavirenz concentrations and clinical correlates in a retrospective cohort of Ghanaian HIV-infected patients. J. Antimicrob. Chemother. 2014, 69, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, L.; Amin, J.; Else, L.; Boffito, M.; Egan, D.; Owen, A.; Khoo, S.; Back, D.; Orrell, C.; Clarke, A.; et al. Comprehensive pharmacokinetic, pharmacodynamic and pharmacogenetic evaluation of once-daily efavirenz 400 and 600 mg in treatment-naive HIV-infected patients at 96 weeks: Results of the ENCORE1 study. Clin. Pharmacokinet. 2016, 55, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Neuhaus, J.; Chu, H.; Neaton, J.; Wyen, C.; Rockstroh, J.K.; Skiest, D.J.; Boyd, M.A.; Khoo, S.; Rotger, M.; et al. Investigation of efavirenz discontinuation in multi-ethnic populations of HIV-positive individuals by genetic analysis. EBioMedicine 2015, 2, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Kiang, T.K.; Ho, P.C.; Anari, M.R.; Tong, V.; Abbott, F.S.; Chang, T.K. Contribution of CYP2C9, CYP2A6, and CYP2B6 to valproic acid metabolism in hepatic microsomes from individuals with the CYP2C9*1/*1 genotype. Toxicol. Sci. 2006, 94, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Stephens, J.R.; Levy, R.H. Valproate hepatotoxicity syndrome: Hypotheses of pathogenesis. Pharm. Weekbl. Sci. 1992, 14, 118–121. [Google Scholar] [PubMed]
- Rettie, A.E.; Rettenmeier, A.W.; Howald, W.N.; Baillie, T.A. Cytochrome P-450—Catalyzed formation of delta 4-VPA, a toxic metabolite of valproic acid. Science 1987, 235, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Sadeque, A.J.; Fisher, M.B.; Korzekwa, K.R.; Gonzalez, F.J.; Rettie, A.E. Human CYP2C9 and CYP2A6 mediate formation of the hepatotoxin 4-ene-valproic acid. J. Pharmacol. Exp. Ther. 1997, 283, 698–703. [Google Scholar] [PubMed]
- Tan, L.; Yu, J.T.; Sun, Y.P.; Ou, J.R.; Song, J.H.; Yu, Y. The influence of cytochrome oxidase CYP2A6, CYP2B6, and CYP2C9 polymorphisms on the plasma concentrations of valproic acid in epileptic patients. Clin. Neurol. Neurosurg. 2010, 112, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, P.; Yang, L.P.; Pan, J.; Yang, X.; Ma, H.Y. Association of CYP2C9, CYP2A6, ACSM2A, and CPT1A gene polymorphisms with adverse effects of valproic acid in Chinese patients with epilepsy. Epilepsy Res. 2017, 132, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Ban, M.; Hirata, K.; Yamamoto, A.; Hara, Y.; Momose, Y. Involvement of CYP2A6 in the formation of a novel metabolite, 3-hydroxypilocarpine, from pilocarpine in human liver microsomes. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Kimonen, T.; Juvonen, R.O.; Alhava, E.; Pasanen, M. The inhibition of CYP enzymes in mouse and human liver by pilocarpine. Br. J. Pharmacol. 1995, 114, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Kinonen, T.; Pasanen, M.; Gynther, J.; Poso, A.; Jarvinen, T.; Alhava, E.; Juvonen, R.O. Competitive inhibition of coumarin 7-hydroxylation by pilocarpine and its interaction with mouse CYP 2A5 and human CYP 2A6. Br. J. Pharmacol. 1995, 116, 2625–2630. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Nakajima, M.; Fukami, T.; Hara, Y.; Hasunuma, T.; Yokoi, T.; Momose, Y. Genetic polymorphisms of CYP2A6 affect the in-vivo pharmacokinetics of pilocarpine. Pharmacogenet. Genom. 2008, 18, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Svensson, U.S.; Ashton, M. Identification of the human cytochrome P450 enzymes involved in the in vitro metabolism of artemisinin. Br. J. Clin. Pharmacol. 1999, 48, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Bjorkman, A.; Andersson, T.B.; Gustafsson, L.L.; Masimirembwa, C.M. Identification of human cytochrome P(450)s that metabolise anti-parasitic drugs and predictions of in vivo drug hepatic clearance from in vitro data. Eur. J. Clin. Pharmacol. 2003, 59, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Yusof, W.; Hua, G.S. Gene, ethnic and gender influences predisposition of adverse drug reactions to artesunate among Malaysians. Toxicol. Mech. Methods 2012, 22, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Phompradit, P.; Muhamad, P.; Cheoymang, A.; Na-Bangchang, K. Preliminary investigation of the contribution of CYP2A6, CYP2B6, and UGT1A9 polymorphisms on artesunate-mefloquine treatment response in Burmese patients with Plasmodium falciparum malaria. Am. J. Trop. Med. Hyg. 2014, 91, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Morooka, S.; Koike, H.; Imanishi, N.; Natsume, Y. SM-12502: A platelet activating factor antagonist. Cardiovasc. Drug Rev. 1995, 13, 105–122. [Google Scholar] [CrossRef]
- Nunoya, K.; Yokoi, Y.; Kimura, K.; Kodama, T.; Funayama, M.; Inoue, K.; Nagashima, K.; Funae, Y.; Shimada, N.; Green, C.; et al. (+)-cis-3,5-dimethyl-2-(3-pyridyl) thiazolidin-4-one hydrochloride (SM-12502) as a novel substrate for cytochrome P450 2A6 in human liver microsomes. J. Pharmacol. Exp. Ther. 1996, 277, 768–774. [Google Scholar] [PubMed]
- Nunoya, K.I.; Yokoi, T.; Kimura, K.; Kainuma, T.; Satoh, K.; Kinoshita, M.; Kamataki, T. A new CYP2A6 gene deletion responsible for the in vivo polymorphic metabolism of (+)-cis-3,5-dimethyl-2-(3-pyridyl)thiazolidin-4-one hydrochloride in humans. J. Pharmacol. Exp. Ther. 1999, 289, 437–442. [Google Scholar] [PubMed]
- Kimura, M.; Yamazaki, H.; Fujieda, M.; Kiyotani, K.; Honda, G.; Saruwatari, J.; Nakagawa, K.; Ishizaki, T.; Kamataki, T. Cyp2a6 is a principal enzyme involved in hydroxylation of 1,7-dimethylxanthine, a main caffeine metabolite, in humans. Drug Metab. Dispos. Biol. Fate Chem. 2005, 33, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, N.; Carrillo, J.A.; Gervasini, G.; Jankovic, S.; Aklillu, E. In vivo evaluation of CYP2A6 and xanthine oxidase enzyme activities in the Serbian population. Eur. J. Clin. Pharmacol. 2010, 66, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Murayama, N.; Shimizu, M.; Kobayashi, K.; Kishimoto, I.; Yamazaki, H. Cytochrome P450 2A6 phenotyping using dietary caffeine salivary metabolite ratios and genotyping using blood on storage cards in Non-smoking japanese volunteers. Drug Metab. Lett. 2017, 10, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Aklillu, E.; Djordjevic, N.; Carrillo, J.A.; Makonnen, E.; Bertilsson, L.; Ingelman-Sundberg, M. High CYP2A6 enzyme activity as measured by a caffeine test and unique distribution of CYP2A6 variant alleles in Ethiopian population. OMICS 2014, 18, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, N.; Carrillo, J.A.; van den Broek, M.P.; Kishikawa, J.; Roh, H.K.; Bertilsson, L.; Aklillu, E. Comparisons of CYP2A6 genotype and enzyme activity between Swedes and Koreans. Drug Metab. Pharmacokinet. 2013, 28, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Kadlubar, S.; Anderson, J.P.; Sweeney, C.; Gross, M.D.; Lang, N.P.; Kadlubar, F.F.; Anderson, K.E. Phenotypic CYP2A6 variation and the risk of pancreatic cancer. JOP 2009, 10, 263–270. [Google Scholar] [PubMed]
- Nowell, S.; Sweeney, C.; Hammons, G.; Kadlubar, F.F.; Lang, N.P. CYP2A6 activity determined by caffeine phenotyping: Association with colorectal cancer risk. Cancer Epidemiol. Biomark. Prev. 2002, 11, 377–383. [Google Scholar]
- Rodriguez-Morato, J.; Robledo, P.; Tanner, J.A.; Boronat, A.; Perez-Mana, C.; Oliver Chen, C.Y.; Tyndale, R.F.; de la Torre, R. CYP2D6 and CYP2A6 biotransform dietary tyrosol into hydroxytyrosol. Food Chem. 2017, 217, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Pancorbo, A.; Cerretani, L.; Bendini, A.; Segura-Carretero, A.; Del Carlo, M.; Gallina-Toschi, T.; Lercker, G.; Compagnone, D.; Fernandez-Gutierrez, A. Evaluation of the antioxidant capacity of individual phenolic compounds in virgin olive oil. J. Agric. Food Chem. 2005, 53, 8918–8925. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| CYP2A6 Genetic Variant | rs ID | CYP2A6 Region | Genetic Impact | Functional Impact on CYP2A6 a | Allele Frequency (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| White | African | Asian | Alaska Native | American Indian | ||||||
| Northern Plains | Southwest | |||||||||
| *1B | N/A | 3′-UTR | 58 bp gene conversion with CYP2A7 | Increased mRNA stability | 28–35 | 11–18 | 26–57 | 65 | 69.7 | 61.6 |
| *1X2A and B | N/A | intron 8 and 5.2–5.6 kb 3′ | CYP2A6 gene duplications | Increased mRNA expression | 0–1.7 | 0 | 0–0.4 | 0 | – | – |
| *2 | rs1801272 | Exon 3 | Nonsynonymous, L160H | Substantially decreased enzyme activity | 1.1–5.3 | 0–1.1 | 0 | 0.4 | 0.3 | 0.6 |
| *4 | N/A | N/A | CYP2A6 gene deletion | No mRNA expression | 0.1–4.2 | 0.5–2.7 | 4.9–24 | 15 | 1.6 | 0.3 |
| *5 | rs5031016 | Exon 9 | Nonsynonymous, G479V | Decreased enzyme activity | 0–0.3 | 0 | 0–1.2 | – | – | – |
| *7 | rs5031017 | Exon 9 | Nonsynonymous, I471T | Decreased enzyme activity | 0–0.3 | 0 | 2.2–13 | 0 | 0 | 0 |
| *9 | rs28399433 | 5′ | Promoter SNP, interrupts TATA box (A>C) | Decreased mRNA expression | 5.2–8.0 | 5.7–9.6 | 16–22 | 8.9 | 11.9 | 20.9 |
| *10 | rs5031017, rs28399468 | Exon 9 | Nonsynonymous, I471T, R485L | Inactive enzyme | 0 | 0 | 0.4–4.3 | 1.9 | – | – |
| *12 | esv2663194 | N/A | Exons 1–2 from CYP2A7, exons 3–9 from CYP2A6, 10 amino acid substitution | Decreased enzyme activity | 0–0.3 | 0–0.4 | 0–0.8 | 0.4 | 0.3 | 0.3 |
| *17 | rs28399454 | Exon 7 | Nonsynonymous, V365M | Substantially decreased enzyme activity | 0 | 7.1–11 | 0 | 0 | 0 | 0 |
| *18 | rs1809810 | Exon 8 | Nonsynonymous, Y392F | Decreased enzyme activity | 1.1–2.1 | 0 | 0–0.5 | – | – | – |
| *20 | N/A | Exon 4 | Two-nucleotide deletion, frame shift, truncated protein | Substantially decreased protein levels | 0 | 1.1–1.7 | 0 | – | – | – |
| *21 | rs6413474 | Exon 9 | Nonsynonymous, K476R | Decreased enzyme activity | 0–2.3 | 0–0.6 | 0–3.4 | – | – | – |
| *23 | rs56256500 | Exon 4 | Nonsynonymous, R203C | Decreased enzyme activity | 0 | 1.1–2.0 | 0 | – | – | – |
| *24 | rs72549435, rs143731390 | Exon 2, 9 | Nonsynonymous, V110L, N438Y | Decreased enzyme activity | 0 | 0.7–2.3 | 0 | – | – | – |
| *25 | rs28399440 | Exon 3 | Nonsynonymous, F118L | Decreased enzyme activity | 0 | 0.5–1.2 | 0 | – | – | – |
| *28 | rs28399463 | Exon 8 | Nonsynonymous, N418D, E419D | Decreased enzyme activity | – | 0.9–2.4 | – | – | – | – |
| *35 | rs143732390 | Exon 9 | Nonsynonymous, N438Y | Decreased enzyme activity | 0 | 2.5–2.9 | 0.5–0.8 | 0 | 0 | 0.3 |
| N/A | rs56113850 | Intron 4 | Non-coding SNP (T>C) | Increased protein expression and enzyme activity; top hit in multiple GWASs of NMR | 56–59 | 39 | 29 | 72 | 68 | 65 |
| N/A | rs113288603 | 5′ | Non-coding SNP (C>T) | Decreased enzyme activity | 9–15 | 12 | 23 | – | – | – |
| N/A | rs12459249 | 3′ | Non-coding SNP (T>C) | Increased enzyme activity | 68 | 69–66 | 41 | – | – | – |
| N/A | rs111645190 | 5′ | Non-coding SNP (G>A) | Decreased enzyme activity; may tag the *17 variant | 0 | 14 | 0 | – | – | – |
| N/A | rs57837628 | 5′ | Non-coding SNP (A>G) | Increased protein expression and enzyme activity | 49–54 | 17 | 29 | 70 | 73 | 58 |
| N/A | rs7260629 | 5′ | Non-coding SNP (T>G) | Increased protein expression and enzyme activity | 69–72 | 71 | 74 | 83 | – | – |
| N/A | rs7259706 | 5′ | Non-coding SNP (C>T) | Increased protein expression and enzyme activity | 69–70 | 73 | 73 | 83 | – | – |
| N/A | rs150298687 | 5′ | Non-coding SNP (T>C) | Increased protein expression and enzyme activity | 58–63 | 46 | 45 | 81 | – | – |
| N/A | rs28399453 | Intron 6 | Non-coding SNP (G>A) | Increased protein expression and enzyme activity | 6–7 | 0 | 0 | 3 | 0 | 0.3 |
| N/A | rs8192733 | 3′-UTR | Non-coding SNP (G>C) | Increased protein expression and enzyme activity | 47–48 | 23 | 51 | 66 | – | – |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanner, J.-A.; Tyndale, R.F. Variation in CYP2A6 Activity and Personalized Medicine. J. Pers. Med. 2017, 7, 18. https://doi.org/10.3390/jpm7040018
Tanner J-A, Tyndale RF. Variation in CYP2A6 Activity and Personalized Medicine. Journal of Personalized Medicine. 2017; 7(4):18. https://doi.org/10.3390/jpm7040018
Chicago/Turabian StyleTanner, Julie-Anne, and Rachel F. Tyndale. 2017. "Variation in CYP2A6 Activity and Personalized Medicine" Journal of Personalized Medicine 7, no. 4: 18. https://doi.org/10.3390/jpm7040018
APA StyleTanner, J.-A., & Tyndale, R. F. (2017). Variation in CYP2A6 Activity and Personalized Medicine. Journal of Personalized Medicine, 7(4), 18. https://doi.org/10.3390/jpm7040018