
The Genome-Wide DNA Methylation Profile of Peripheral Blood Is Not Systematically Changed by Short-Time Storage at Room Temperature

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. High Quality Sequencing Data Generated from Time Points up to Three Days
2.2. No Storage-Dependent Change in Canonical Methylation Patterns
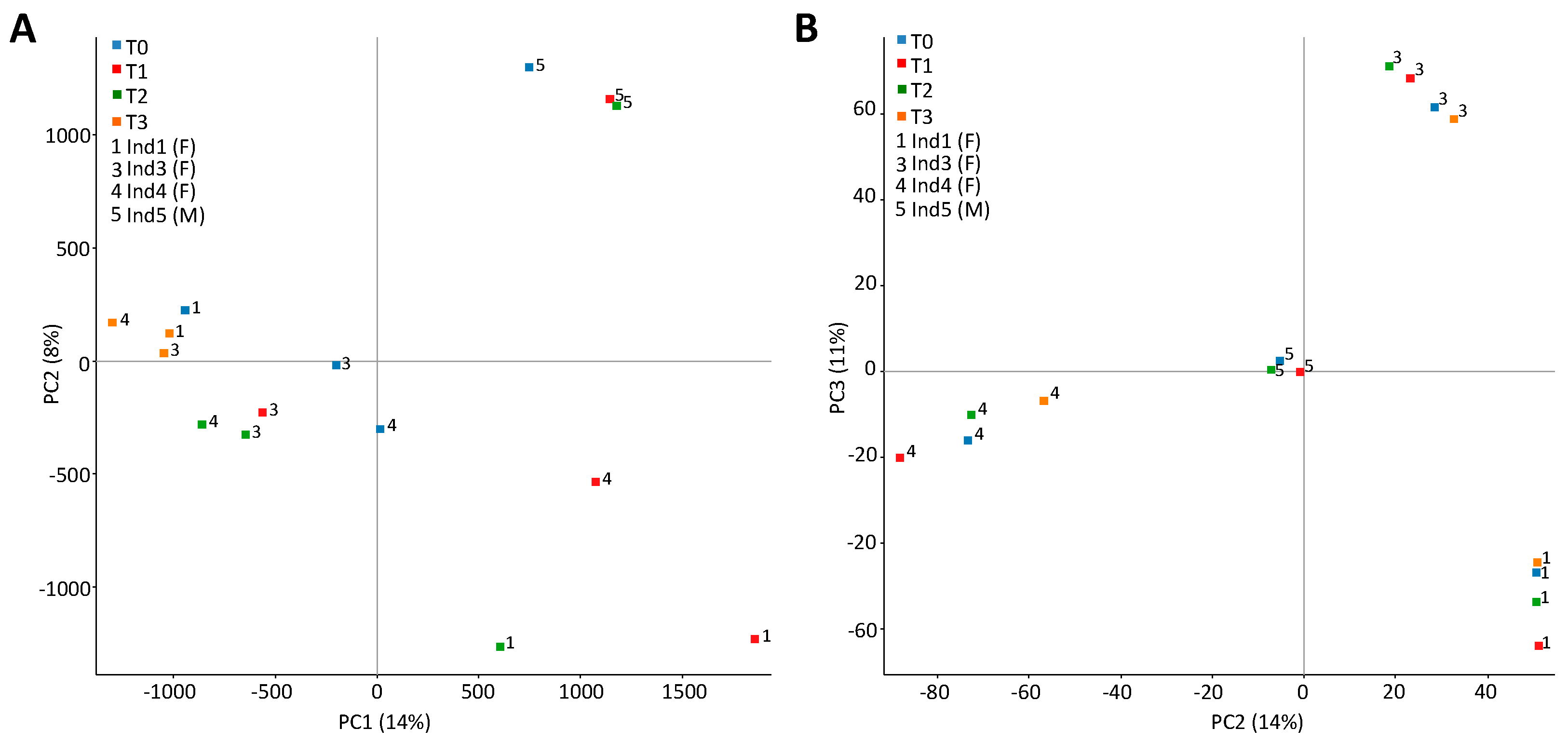
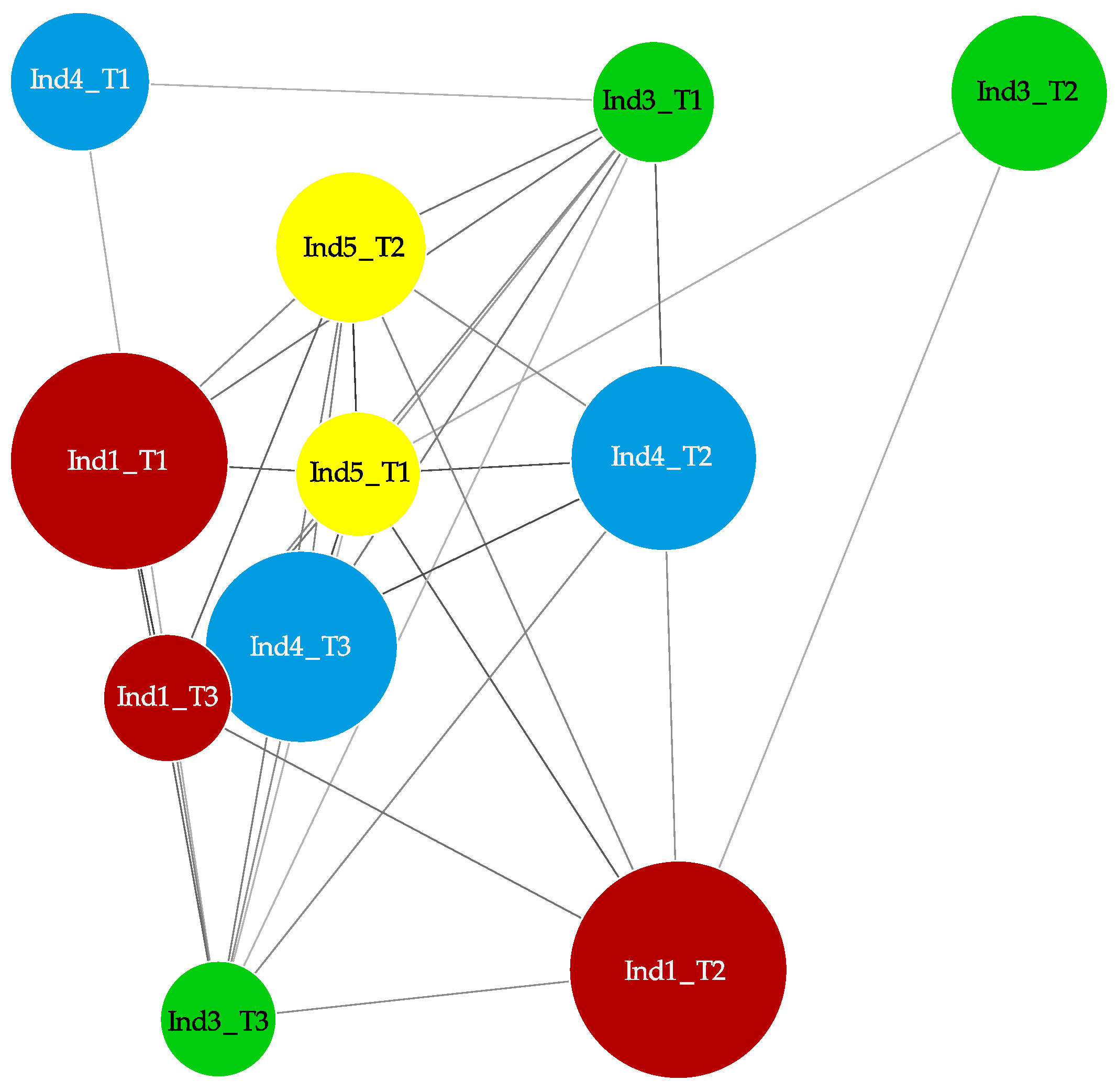
2.3. Stochastic Variation between Time-Points and No Differently Methylated Regions between Groups
3. Discussion
4. Materials and Methods
4.1. Blood Samples
4.2. MeDIP-seq
4.3. Sequencing and Bioinformatics
4.4. Pyrosequencing
4.5. Ethics Statement
4.6. Availability of Data and Materials
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baccarelli, A.; Wright, R.; Bollati, V.; Litonjua, A.; Zanobetti, A.; Tarantini, L.; Sparrow, D.; Vokonas, P.; Schwartz, J. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology 2010, 21, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Lange, N.E.; Sordillo, J.; Tarantini, L.; Bollati, V.; Sparrow, D.; Vokonas, P.; Zanobetti, A.; Schwartz, J.; Baccarelli, A.; Litonjua, A.A.; et al. Alu and LINE-1 methylation and lung function in the normative ageing study. BMJ Open 2012, 2, e001231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosavi, A.; Ardekani, A. Role of Epigenetics in Biology and Human Diseases. Iran. Biomed. J. 2016, 20, 246–258. [Google Scholar] [PubMed]
- Tang, Q.; Cheng, J.; Cao, X.; Surowy, H.; Burwinkel, B. Blood-based DNA methylation as biomarker for breast cancer: A systematic review. Clin. Epigenetics 2016, 8, 115. [Google Scholar] [CrossRef] [PubMed]
- Deneberg, S.; Grovdal, M.; Karimi, M.; Jansson, M.; Nahi, H.; Corbacioglu, A.; Gaidzik, V.; Dohner, K.; Paul, C.; Ekstrom, T.J.; et al. Gene-specific and global methylation patterns predict outcome in patients with acute myeloid leukemia. Leukemia 2010, 24, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Darby, M.M.; Sabunciyan, S. Repetitive elements and epigenetic marks in behavior and psychiatric disease. Adv. Genet. 2014, 86, 185–252. [Google Scholar] [PubMed]
- Joubert, B.R.; Felix, J.F.; Yousefi, P.; Bakulski, K.M.; Just, A.C.; Breton, C.; Reese, S.E.; Markunas, C.A.; Richmond, R.C.; Xu, C.J.; et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016, 98, 680–696. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Petkovich, D.A.; Podolskiy, D.I.; Lobanov, A.V.; Lee, S.G.; Miller, R.A.; Gladyshev, V.N. Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions. Cell Metab. 2017, 25, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Csoka, A.B.; Szyf, M. Epigenetic side-effects of common pharmaceuticals: A potential new field in medicine and pharmacology. Med. Hypotheses 2009, 73, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Staunstrup, N.H.; Starnawska, A.; Nyegaard, M.; Christiansen, L.; Nielsen, A.L.; Borglum, A.; Mors, O. Genome-wide DNA methylation profiling with MeDIP-seq using archived dried blood spots. Clin. Epigenetics 2016, 8, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberg, K.A.; Xie, L.Y.; Nerella, S.; Copeland, W.E.; Costello, E.J.; van den Oord, E.J. High quality methylome-wide investigations through next-generation sequencing of DNA from a single archived dry blood spot. Epigenetics 2013, 8, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Beyan, H.; Down, T.A.; Ramagopalan, S.V.; Uvebrant, K.; Nilsson, A.; Holland, M.L.; Gemma, C.; Giovannoni, G.; Boehm, B.O.; Ebers, G.C.; et al. Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res. 2012, 22, 2138–2145. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, M.N.; Oshlack, A.; Theda, C.; Davis, P.G.; Martino, D.; Sheehan, P.; Dai, Y.; Saffery, R.; Doyle, L.W.; Craig, J.M. Analysis of epigenetic changes in survivors of preterm birth reveals the effect of gestational age and evidence for a long term legacy. Genome Med. 2013, 5, 96. [Google Scholar] [CrossRef] [PubMed]
- Hollegaard, M.V.; Grauholm, J.; Norgaard-Pedersen, B.; Hougaard, D.M. DNA methylome profiling using neonatal dried blood spot samples: A proof-of-principle study. Mol. Genet. Metab. 2013, 108, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.E.; Wong, E.M.; Baglietto, L.; Jung, C.H.; Tsimiklis, H.; Park, D.J.; Wong, N.C.; English, D.R.; Hopper, J.L.; Severi, G.; et al. The use of DNA from archival dried blood spots with the Infinium HumanMethylation450 array. BMC Biotechnol. 2013, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Bulla, A.; De Witt, B.; Ammerlaan, W.; Betsou, F.; Lescuyer, P. Blood DNA Yield but Not Integrity or Methylation Is Impacted After Long-Term Storage. Biopreserv. Biobank. 2016, 14, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Malentacchi, F.; Ciniselli, C.M.; Pazzagli, M.; Verderio, P.; Barraud, L.; Hartmann, C.C.; Pizzamiglio, S.; Weisbuch, S.; Wyrich, R.; Gelmini, S. Influence of pre-analytical procedures on genomic DNA integrity in blood samples: The SPIDIA experience. Clin. Chim. Acta 2015, 440, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Buesa, C.; Maes, T.; Subirada, F.; Barrachina, M.; Ferrer, I. DNA chip technology in brain banks: Confronting a degrading world. J. Neuropathol. Exp. Neurol. 2004, 63, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Santpere, G.; Arzberger, T.; Bell, J.; Blanco, R.; Boluda, S.; Budka, H.; Carmona, M.; Giaccone, G.; Krebs, B.; et al. Brain protein preservation largely depends on the postmortem storage temperature: Implications for study of proteins in human neurologic diseases and management of brain banks: A BrainNet Europe Study. J. Neuropathol. Exp. Neurol. 2007, 66, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Avila, L.; Yuen, R.K.; Diego-Alvarez, D.; Penaherrera, M.S.; Jiang, R.; Robinson, W.P. Evaluating DNA methylation and gene expression variability in the human term placenta. Placenta 2010, 31, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Barres, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-CpG methylation of the PGC-1α promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Barres, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O'Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef] [PubMed]
- McCullumsmith, R.E.; Meador-Woodruff, J.H. Novel approaches to the study of postmortem brain in psychiatric illness: Old limitations and new challenges. Biol. Psychiatry 2011, 69, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, H.T.; Sigurdsson, M.I.; Fallin, M.D.; Irizarry, R.A.; Aspelund, T.; Cui, H.; Yu, W.; Rongione, M.A.; Ekstrom, T.J.; Harris, T.B.; et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA 2008, 299, 2877–2883. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Morgan, M.; Hutchison, K.; Calhoun, V.D. A study of the influence of sex on genome wide methylation. PLoS ONE 2010, 5, e10028. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.E.; Irizarry, R.A. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014, 15, R31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Jones, M.J.; Chen, E.; Neumann, S.M.; Fraser, H.B.; Miller, G.E.; Kobor, M.S. Discordance of DNA methylation variance between two accessible human tissues. Sci. Rep. 2015, 5, 8257. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.M.; Siegmund, K.D.; Pan, F.; Weisenberger, D.J.; Kanel, G.; Laird, P.W.; Yang, A.S. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum. Mol. Genet. 2009, 18, 4808–4817. [Google Scholar] [CrossRef] [PubMed]
- Lokk, K.; Modhukur, V.; Rajashekar, B.; Martens, K.; Magi, R.; Kolde, R.; Koltsina, M.; Nilsson, T.K.; Vilo, J.; Salumets, A.; et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014, 15, r54. [Google Scholar] [CrossRef] [PubMed]
- Solyom, S.; Kazazian, H.H., Jr. Mobile elements in the human genome: Implications for disease. Genome Med. 2012, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Martinez, A.; Boluda, S.; Parchi, P.; Barrachina, M. Brain banks: Benefits, limitations and cautions concerning the use of post-mortem brain tissue for molecular studies. Cell Tissue Bank. 2008, 9, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Vilahur, N.; Baccarelli, A.A.; Bustamante, M.; Agramunt, S.; Byun, H.M.; Fernandez, M.F.; Sunyer, J.; Estivill, X. Storage conditions and stability of global DNA methylation in placental tissue. Epigenomics 2013, 5, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Wockner, L.F.; Noble, E.P.; Lawford, B.R.; Young, R.M.; Morris, C.P.; Whitehall, V.L.; Voisey, J. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry 2014, 4, e339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, A.; Heredia, N.; Senut, M.C.; Hess, M.; Land, S.; Qu, W.; Hollacher, K.; Dereski, M.O.; Ruden, D.M. Early life lead exposure causes gender-specific changes in the DNA methylation profile of DNA extracted from dried blood spots. Epigenomics 2015, 7, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Dugue, P.A.; English, D.R.; MacInnis, R.J.; Jung, C.H.; Bassett, J.K.; FitzGerald, L.M.; Wong, E.M.; Joo, J.E.; Hopper, J.L.; Southey, M.C.; et al. Reliability of DNA methylation measures from dried blood spots and mononuclear cells using the HumanMethylation450k BeadArray. Sci. Rep. 2016, 6, 30317. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Qiu, C.; Li, J.; Tian, Q.; Deng, H.W. Characterization of the DNA methylome and its interindividual variation in human peripheral blood monocytes. Epigenomics 2013, 5, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Antwih, D.A.; Gabbara, K.M.; Lancaster, W.D.; Ruden, D.M.; Zielske, S.P. Radiation-induced epigenetic DNA methylation modification of radiation-response pathways. Epigenetics 2013, 8, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Lienhard, M.; Grimm, C.; Morkel, M.; Herwig, R.; Chavez, L. MEDIPS: Genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 2014, 30, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Statham, A.L.; Strbenac, D.; Coolen, M.W.; Stirzaker, C.; Clark, S.J.; Robinson, M.D. Repitools: An R package for the analysis of enrichment-based epigenomic data. Bioinformatics 2010, 26, 1662–1663. [Google Scholar] [CrossRef] [PubMed]
- Lienhard, M.; Grasse, S.; Rolff, J.; Frese, S.; Schirmer, U.; Becker, M.; Borno, S.; Timmermann, B.; Chavez, L.; Sultmann, H.; et al. QSEA-modelling of genome-wide DNA methylation from sequencing enrichment experiments. Nucleic Acids Res. 2017, 45, e44. [Google Scholar] [CrossRef] [PubMed]
- Halachev, K.; Bast, H.; Albrecht, F.; Lengauer, T.; Bock, C. EpiExplorer: Live exploration and global analysis of large epigenomic datasets. Genome Biol. 2012, 13, R96. [Google Scholar] [CrossRef] [PubMed]
- Fa, S.; Larsen, T.V.; Bilde, K.; Daugaard, T.F.; Ernst, E.H.; Olesen, R.H.; Mamsen, L.S.; Ernst, E.; Larsen, A.; Nielsen, A.L. Assessment of global DNA methylation in the first trimester fetal tissues exposed to maternal cigarette smoking. Clin. Epigenetics 2016, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Worswick, S.; Byun, H.M.; Shear, T.; Soussa, J.C.; Wolff, E.M.; Douer, D.; Garcia-Manero, G.; Liang, G.; Yang, A.S. Changes in DNA methylation of tandem DNA repeats are different from interspersed repeats in cancer. Int. J. Cancer 2009, 125, 723–729. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staunstrup, N.H.; Starnawska, A.; Nyegaard, M.; Nielsen, A.L.; Børglum, A.D.; Mors, O. The Genome-Wide DNA Methylation Profile of Peripheral Blood Is Not Systematically Changed by Short-Time Storage at Room Temperature. Epigenomes 2017, 1, 23. https://doi.org/10.3390/epigenomes1030023
Staunstrup NH, Starnawska A, Nyegaard M, Nielsen AL, Børglum AD, Mors O. The Genome-Wide DNA Methylation Profile of Peripheral Blood Is Not Systematically Changed by Short-Time Storage at Room Temperature. Epigenomes. 2017; 1(3):23. https://doi.org/10.3390/epigenomes1030023
Chicago/Turabian StyleStaunstrup, Nicklas Heine, Anna Starnawska, Mette Nyegaard, Anders Lade Nielsen, Anders Dupont Børglum, and Ole Mors. 2017. "The Genome-Wide DNA Methylation Profile of Peripheral Blood Is Not Systematically Changed by Short-Time Storage at Room Temperature" Epigenomes 1, no. 3: 23. https://doi.org/10.3390/epigenomes1030023
APA StyleStaunstrup, N. H., Starnawska, A., Nyegaard, M., Nielsen, A. L., Børglum, A. D., & Mors, O. (2017). The Genome-Wide DNA Methylation Profile of Peripheral Blood Is Not Systematically Changed by Short-Time Storage at Room Temperature. Epigenomes, 1(3), 23. https://doi.org/10.3390/epigenomes1030023