Targeting EZH2 in Multiple Myeloma—Multifaceted Anti-Tumor Activity

Abstract
:1. Introduction
2. Deregulation of Chromatin Regulators in Multiple Myeloma
3. EZH2 in Multiple Myeloma
4. EZH2 Inhibition in Multiple Myeloma
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Waddington, C.H. The epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Yamada, Y.; Yamanaka, S. Epigenetic regulation in pluripotent stem cells: A key to breaking the epigenetic barrier. Philos. Trans. R. Soc. B. 2013, 368, 20120292. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, R.D.; Morey, R.; Laurent, L.C. The epigenome in pluripotency and differentiation. Epigenomics 2014, 6, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sabari, B.R.; Garcia, B.A.; Allis, C.D.; Zhao, Y. Snapshot: Histone modifications. Cell 2014, 159, 458–458. [Google Scholar] [CrossRef] [PubMed]
- Verdone, L.; Caserta, M.; di Mauro, E. Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 2005, 83, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Litt, M.; Qiu, Y.; Huang, S. Histone arginine methylations: Their roles in chromatin dynamics and transcriptional regulation. Biosci. Rep. 2009, 29, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, A.; Bedford, M.T. Histone arginine methylation. FEBS Lett. 2011, 585, 2024–2031. [Google Scholar] [CrossRef] [PubMed]
- Latham, J.A.; Dent, S.Y. Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 2007, 14, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Lennartsson, A.; Ekwall, K. Histone modification patterns and epigenetic codes. Biochim. Biophys. Acta 2009, 1790, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, T.B.; Veland, N.; Chen, T. Writers, Readers, and Erasers of Epigenetic Marks. In Epigenetic Cancer Therapy; Gray, S.G., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 31–66. [Google Scholar]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J., 3rd. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Wardell, C.P.; Chiecchio, L.; Smith, E.M.; Boyd, K.D.; Neri, A.; Davies, F.E.; Ross, F.M.; Morgan, G.J. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011, 117, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Brioli, A.; Johnson, D.C.; Kaiser, M.F.; Mirabella, F.; Lopez-Corral, L.; Humphray, S.; Murray, L.; et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia 2014, 28, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Podar, K.; Breitkreutz, I.; Richardson, P.G.; Anderson, K.C. Multiple myeloma. Lancet 2009, 374, 324–339. [Google Scholar] [CrossRef]
- Kumar, S. Multiple myeloma—Current issues and controversies. Cancer Treat. Rev. 2010, 36, S3–S11. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M. Chromosome translocations in multiple myeloma. Oncogene 2001, 20, 5611–5622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsagel, P.L.; Kuehl, W.M. Molecular pathogenesis and a consequent classification of multiple myeloma. J. Clin. Oncol. 2005, 23, 6333–6338. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broyl, A.; Hose, D.; Lokhorst, H.; de Knegt, Y.; Peeters, J.; Jauch, A.; Bertsch, U.; Buijs, A.; Stevens-Kroef, M.; Beverloo, H.B.; et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood 2010, 116, 2543–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; van Ness, B.; Chesi, M.; Minvielle, S.; et al. International myeloma working group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.M.; Chiecchio, L.; Dagrada, G.; Protheroe, R.K.; Stockley, D.M.; Harrison, C.J.; Cross, N.C.; Szubert, A.J.; Drayson, M.T.; Morgan, G.J. The t(14;20) is a poor prognostic factor in myeloma but is associated with long–term stable disease in monoclonal gammopathies of undetermined significance. Haematologica 2010, 95, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Joao, C.; Costa, C.; Coelho, I.; Vergueiro, M.J.; Ferreira, M.; da Silva, M.G. Long-term survival in multiple myeloma. Clin. Case Rep. 2014, 2, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, T.V.; Kumar, S.K.; Srivastava, G.; Gertz, M.A.; Lacy, M.Q.; Buadi, F.K.; Kyle, R.A.; Dispenzieri, A. Long–term response to lenalidomide in patients with newly diagnosed multiple myeloma. Leukemia 2014, 28, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Hideshima, T.; Anderson, K.C. Novel therapeutic strategies for multiple myeloma. Exp. Hematol. 2015, 43, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlyn, C.; Kaiser, M.; Heuck, C.; Melchor, L.; Wardell, C.; Murison, A.; Chavan, S.; Johnson, D.C.; Begum, D.B.; Dahir, N.; et al. The spectrum and clinical impact of epigenetic modifier mutations in myeloma. Clin. Cancer Res. 2016, 22, 5783–5794. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K.; Fonseca, R. Prognostic and therapeutic significance of myeloma genetics and gene expression profiling. J. Clin. Oncol. 2005, 23, 6339–6344. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, K.; Gimsing, P.; Gronbaek, K. The role of epigenetics in the biology of multiple myeloma. Blood Cancer J. 2014, 4, e207. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol 2016, 104, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Dupere-Richer, D.; Licht, J.D. Epigenetic regulatory mutations and epigenetic therapy for multiple myeloma. Curr. Opin. Hematol. 2017, 24, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Alzrigat, M.; Párraga, A.A.; Jernberg-Wiklund, H. Epigenetics in multiple myeloma: From mechanisms to therapy. Semin. Cancer Biol. 2018, 51, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational spectrum, copy number changes, and outcome: Results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Lagana, A.; Perumal, D.; Melnekoff, D.; Readhead, B.; Kidd, B.A.; Leshchenko, V.; Kuo, P.Y.; Keats, J.; DeRome, M.; Yesil, J.; et al. Integrative network analysis identifies novel drivers of pathogenesis and progression in newly diagnosed multiple myeloma. Leukemia 2018, 32, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Jagani, Z.; Wiederschain, D.; Loo, A.; He, D.; Mosher, R.; Fordjour, P.; Monahan, J.; Morrissey, M.; Yao, Y.M.; Lengauer, C.; et al. The polycomb group protein bmi–1 is essential for the growth of multiple myeloma cells. Cancer Res. 2010, 70, 5528–5538. [Google Scholar] [CrossRef] [PubMed]
- Bolomsky, A.; Schlangen, K.; Schreiner, W.; Zojer, N.; Ludwig, H. Targeting of BMI-1 with PTC-209 shows potent anti-myeloma activity and impairs the tumor microenvironment. J. Hematol. Oncol. 2016, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Reiman, T.; Maxwell, C.A.; Taylor, B.J.; Larratt, L.M.; Mant, M.J.; Belch, A.R.; Pilarski, L.M. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 2003, 101, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Maxwell, C.A.; Taylor, B.J.; Hendzel, M.J.; Chesi, M.; Bergsagel, P.L.; Larratt, L.M.; Mant, M.J.; Reiman, T.; Belch, A.R.; et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood 2005, 105, 4060–4069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Garcia, E.; Popovic, R.; Min, D.J.; Sweet, S.M.; Thomas, P.M.; Zamdborg, L.; Heffner, A.; Will, C.; Lamy, L.; Staudt, L.M.; et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011, 117, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Popovic, R.; Martinez-Garcia, E.; Giannopoulou, E.G.; Zhang, Q.; Ezponda, T.; Shah, M.Y.; Zheng, Y.; Will, C.M.; Small, E.C.; Hua, Y.; et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014, 10, e1004566. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.Y.; Martinez-Garcia, E.; Phillip, J.M.; Chambliss, A.B.; Popovic, R.; Ezponda, T.; Small, E.C.; Will, C.; Phillip, M.P.; Neri, P.; et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 2016, 35, 5905–5915. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.R.; Tian, E.; Heuck, C.J.; Johann, D.J.; Epstein, J.; Swanson, C.M.; Lukacs, J.L.; Binz, R.L.; Johnson, M.; Sammartino, G.; et al. Evidence of an epigenetic origin for high-risk 1q21 copy number aberrations in multiple myeloma. Blood 2015, 125, 3756–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Chen, L.; Barlogie, B.; Stephens, O.; Wu, X.; Williams, D.R.; Cartron, M.A.; van Rhee, F.; Nair, B.; Waheed, S.; et al. High-risk myeloma is associated with global elevation of miRNAs and overexpression of EIF2C2/AGO2. Proc. Natl. Acad. Sci. USA 2010, 107, 7904–7909. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Biasiolo, M.; Agnelli, L.; Todoerti, K.; Mosca, L.; Fabris, S.; Sales, G.; Deliliers, G.L.; Bicciato, S.; Lombardi, L.; et al. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood 2009, 114, e20–e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, J.; Ballabio, E.; Chen, X.H.; Kusec, R.; Taylor, S.; Hay, D.; Tramonti, D.; Saunders, N.J.; Littlewood, T.; Pezzella, F.; et al. MicroRNA expression in multiple myeloma is associated with genetic subtype, isotype and survival. Biol. Direct 2011, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Boyd, K.; Davies, F.E. The potential role of epigenetic therapy in multiple myeloma. Br. J. Haematol. 2010, 148, 702–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, K.; Menu, E.; van Valckenborgh, E.; van Riet, I.; Vanderkerken, K.; de Bruyne, E. Epigenetic modulating agents as a new therapeutic approach in multiple myeloma. Cancers 2013, 5, 430–461. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.E.; Takhsha, F.S.; Chirumamilla, C.S.; Perez-Novo, C.; Vanden Berghe, W.; Cuendet, M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenetics 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Richardson, P.G.; Anderson, K.C. Promising therapies in multiple myeloma. Blood 2015, 126, 300–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the panorama 1 trial): A randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 2016, 3, e506–e515. [Google Scholar] [CrossRef]
- Richardson, P.G.; Harvey, R.D.; Laubach, J.P.; Moreau, P.; Lonial, S.; San-Miguel, J.F. Panobinostat for the treatment of relapsed or relapsed/refractory multiple myeloma: Pharmacology and clinical outcomes. Expert Rev. Clin. Pharmacol. 2016, 9, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Wahaib, K.; Beggs, A.E.; Campbell, H.; Kodali, L.; Ford, P.D. Panobinostat: A histone deacetylase inhibitor for the treatment of relapsed or refractory multiple myeloma. Am. J. Health Syst. Pharm. 2016, 73, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument–Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in polycomb–group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Di Croce, L.; Helin, K. Transcriptional regulation by polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Konze, K.D.; Jin, J.; Wang, G.G. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp. Hematol. 2015, 43, 698–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnick, A. Epigenetic therapy leaps ahead with specific targeting of EZH2. Cancer Cell 2012, 22, 569–570. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet. Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Chang, C.J.; Hung, M.C. The role of EZH2 in tumour progression. Br. J. Cancer 2012, 106, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Ezponda, T.; Licht, J.D. Molecular pathways: Deregulation of histone H3 lysine 27 methylation in cancer-different paths, same destination. Clin. Cancer Res. 2014, 20, 5001–5008. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Konze, K.D.; Tao, J. Polycomb genes, miRNA, and their deregulation in B-cell malignancies. Blood 2015, 125, 1217–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herviou, L.; Cavalli, G.; Cartron, G.; Klein, B.; Moreaux, J. EZH2 in normal hematopoiesis and hematological malignancies. Oncotarget 2016, 7, 2284–2296. [Google Scholar] [CrossRef] [PubMed]
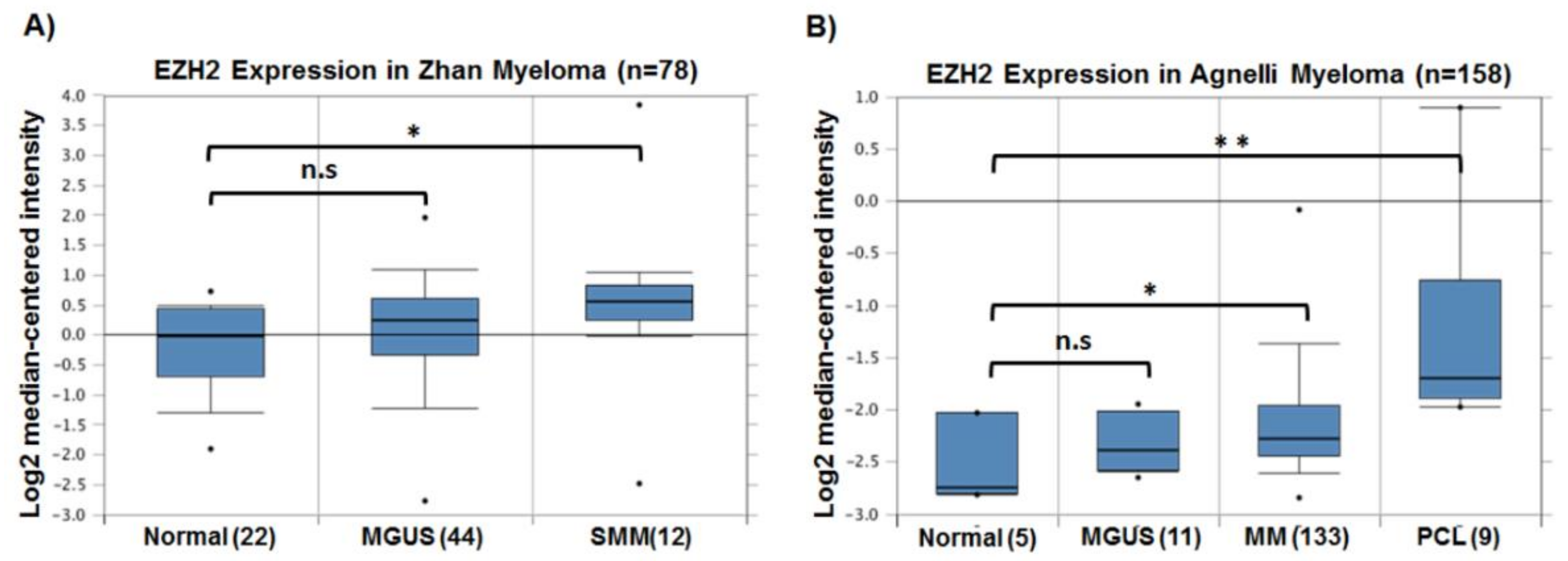
- Zhan, F.; Hardin, J.; Kordsmeier, B.; Bumm, K.; Zheng, M.; Tian, E.; Sanderson, R.; Yang, Y.; Wilson, C.; Zangari, M.; et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood 2002, 99, 1745–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, F.; Tian, E.; Bumm, K.; Smith, R.; Barlogie, B.; Shaughnessy, J., Jr. Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late-stage B-cell development. Blood 2003, 101, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
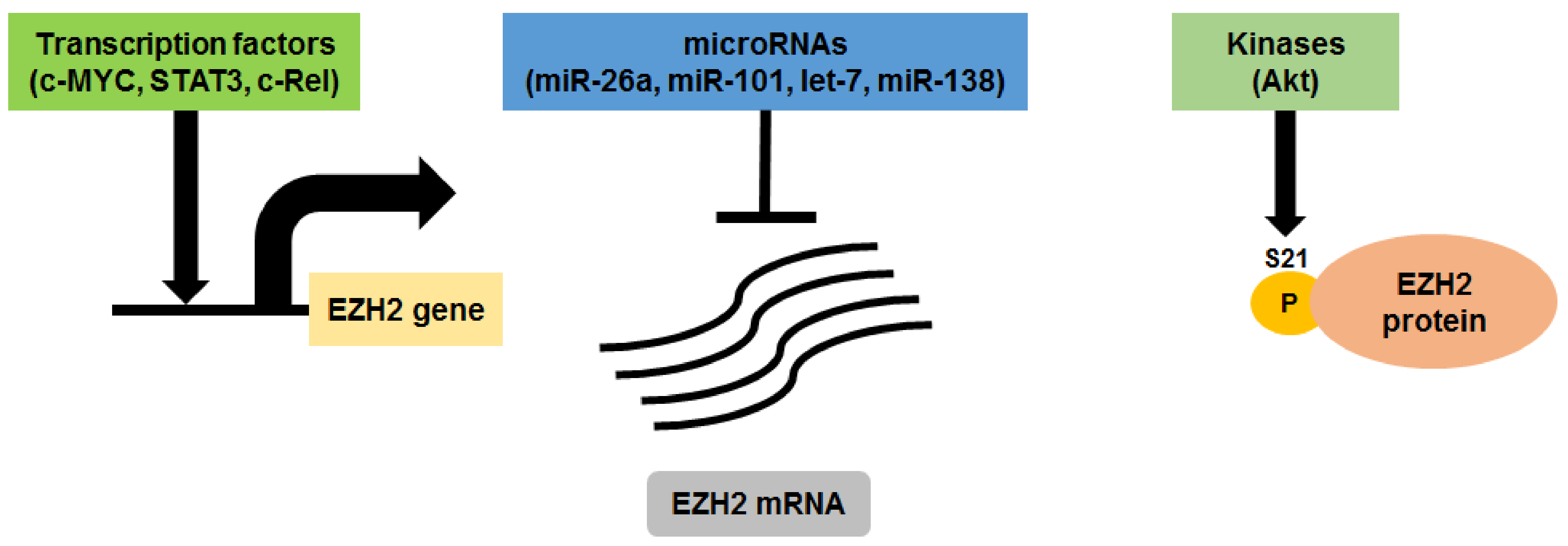
- Croonquist, P.A.; van Ness, B. The polycomb group protein enhancer of zeste homolog 2 (EZH2) is an oncogene that influences myeloma cell growth and the mutant RAS phenotype. Oncogene 2005, 24, 6269–6280. [Google Scholar] [CrossRef] [PubMed]
- Kalushkova, A.; Fryknas, M.; Lemaire, M.; Fristedt, C.; Agarwal, P.; Eriksson, M.; Deleu, S.; Atadja, P.; Osterborg, A.; Nilsson, K.; et al. Polycomb target genes are silenced in multiple myeloma. PLoS ONE 2010, 5, e11483. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Bright, M.D.; Buros, A.F.; Stein, C.K.; Walters, Z.; Aronson, L.I.; Mirabella, F.; Jones, J.R.; Kaiser, M.F.; Walker, B.A.; et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017, 7, e549. [Google Scholar] [CrossRef] [PubMed]
- Neo, W.H.; Lim, J.F.; Grumont, R.; Gerondakis, S.; Su, I.H. c-Rel regulates EZH2 expression in activated lymphocytes and malignant lymphoid cells. J. Biol. Chem. 2014, 289, 31693–31707. [Google Scholar] [CrossRef] [PubMed]
- Pichiorri, F.; Suh, S.S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.P.; et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 12885–12890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seckinger, A.; Meissner, T.; Moreaux, J.; Benes, V.; Hillengass, J.; Castoldi, M.; Zimmermann, J.; Ho, A.D.; Jauch, A.; Goldschmidt, H.; et al. miRNAs in multiple myeloma—A survival relevant complex regulator of gene expression. Oncotarget 2015, 6, 39165–39183. [Google Scholar] [CrossRef] [PubMed]
- Rastgoo, N.; Pourabdollah, M.; Abdi, J.; Reece, D.; Chang, H. Dysregulation of EZH2/miR-138 axis contributes to drug resistance in multiple myeloma by downregulating RBPMS. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Koyama, D.; Wada, T.; Izumi, T.; Hofgaard, P.O.; Bogen, B.; Furukawa, Y. Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J. Clin. Invest. 2015, 125, 4375–4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Sneeringer, C.J.; Scott, M.P.; Kuntz, K.W.; Knutson, S.K.; Pollock, R.M.; Richon, V.M.; Copeland, R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone h3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 20980–20985. [Google Scholar] [CrossRef] [PubMed]
- Majer, C.R.; Jin, L.; Scott, M.P.; Knutson, S.K.; Kuntz, K.W.; Keilhack, H.; Smith, J.J.; Moyer, M.P.; Richon, V.M.; Copeland, R.A.; et al. A687V EZH2 is a gain-of-function mutation found in lymphoma patients. FEBS Lett. 2012, 586, 3448–3451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Tsirigos, A.; van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres–Marco, D.; da Ros, V.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in t cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Nikoloski, G.; Langemeijer, S.M.; Kuiper, R.P.; Knops, R.; Massop, M.; Tonnissen, E.R.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. EZH2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat. Commun. 2014, 5, 4177. [Google Scholar] [CrossRef] [PubMed]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Alzrigat, M.; Parraga, A.A.; Enroth, S.; Singh, U.; Ungerstedt, J.; Osterborg, A.; Brown, P.J.; Ma, A.; Jin, J.; et al. Genome-wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget 2016, 7, 6809–6823. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. Oncomine: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Zhan, F.; Barlogie, B.; Arzoumanian, V.; Huang, Y.; Williams, D.R.; Hollmig, K.; Pineda-Roman, M.; Tricot, G.; van Rhee, F.; Zangari, M.; et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood 2007, 109, 1692–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnelli, L.; Mosca, L.; Fabris, S.; Lionetti, M.; Andronache, A.; Kwee, I.; Todoerti, K.; Verdelli, D.; Battaglia, C.; Bertoni, F.; et al. A SNP microarray and fish-based procedure to detect allelic imbalances in multiple myeloma: An integrated genomics approach reveals a wide gene dosage effect. Genes Chromosomes Cancer 2009, 48, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.J.; Cheung, P.; Chen, K.; Zee, B.M.; Kioi, M.; Lauring, J.; Xi, Y.; Park, B.H.; Shi, X.; Garcia, B.A.; et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 2011, 44, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Ezponda, T.; Dupere-Richer, D.; Will, C.M.; Small, E.C.; Varghese, N.; Patel, T.; Nabet, B.; Popovic, R.; Oyer, J.; Bulic, M.; et al. UTX/KDM6A loss enhances the malignant phenotype of multiple myeloma and sensitizes cells to EZH2 inhibition. Cell Rep. 2017, 21, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Rius, M.; Lyko, F. Epigenetic cancer therapy: Rationales, targets and drugs. Oncogene 2012, 31, 4257–4265. [Google Scholar] [CrossRef] [PubMed]
- Gaudichon, J.; Milano, F.; Cahu, J.; DaCosta, L.; Martens, A.C.; Renoir, J.M.; Sola, B. Deazaneplanocin a is a promising drug to kill multiple myeloma cells in their niche. PLoS ONE 2014, 9, e107009. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bi, C.; Cheong, L.L.; Liu, S.C.; Huang, G.; Zhou, J.; Yu, Q.; Chen, C.S.; Chng, W.J. Determinants of sensitivity to DZNep induced apoptosis in multiple myeloma cells. PLoS ONE 2011, 6, e21583. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Bahlis, N.J.; Lonial, S. Panobinostat for the treatment of multiple myeloma. Expert Opin. Investig. Drugs 2012, 21, 733–747. [Google Scholar] [CrossRef] [PubMed]
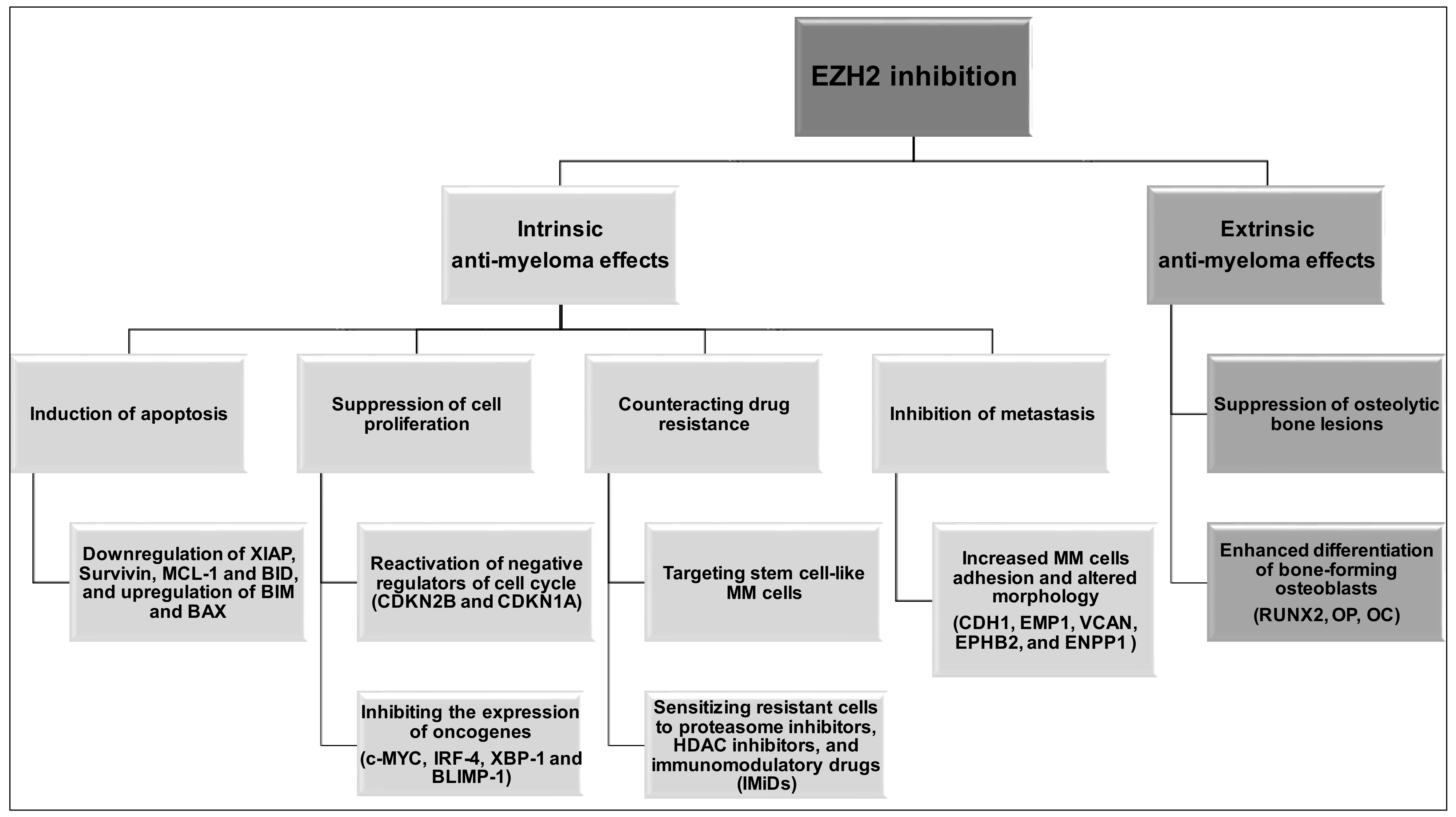
- Alzrigat, M.; Parraga, A.A.; Agarwal, P.; Zureigat, H.; Osterborg, A.; Nahi, H.; Ma, A.; Jin, J.; Nilsson, K.; Oberg, F.; et al. EZH2 inhibition in multiple myeloma downregulates myeloma associated oncogenes and upregulates microRNAs with potential tumor suppressor functions. Oncotarget 2017, 8, 10213–10224. [Google Scholar] [CrossRef] [PubMed]
- Rizq, O.; Mimura, N.; Oshima, M.; Saraya, A.; Koide, S.; Kato, Y.; Aoyama, K.; Nakajima-Takagi, Y.; Wang, C.; Chiba, T.; et al. Dual inhibition of EZH2 and EZH1 sensitizes PRC2-dependent tumors to proteasome inhibition. Clin. Cancer Res. 2017, 23, 4817–4830. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Liu, M.; Pan, J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells. Oncotarget 2017, 8, 3396–3411. [Google Scholar] [CrossRef] [PubMed]
- Harding, T.; Swanson, J.; van Ness, B. EZH2 inhibitors sensitize myeloma cell lines to panobinostat resulting in unique combinatorial transcriptomic changes. Oncotarget 2018, 9, 21930–21942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, K.; Sogaard Helbo, A.; Fibiger Munch-Petersen, H.; Sjo, L.; Christensen, J.; Sommer Kristensen, L.; Asmar, F.; Hermansen, N.E.U.; O’Connel, C.; Gimsing, P.; et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol. Oncol. 2018, 12, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Hernando, H.; Gelato, K.A.; Lesche, R.; Beckmann, G.; Koehr, S.; Otto, S.; Steigemann, P.; Stresemann, C. EZH2 inhibition blocks multiple myeloma cell growth through upregulation of epithelial tumor suppressor genes. Mol. Cancer Ther. 2016, 15, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Honma, D.; Kanno, O.; Watanabe, J.; Kinoshita, J.; Hirasawa, M.; Nosaka, E.; Shiroishi, M.; Takizawa, T.; Yasumatsu, I.; Horiuchi, T.; et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017, 108, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Johnson, P.W.M.; Winter, J.; Leonard, J.; Giulino-Roth, L.; Horner, T.; Radswillas, K.; Carver, J.; Dhar, A. A phase I, open-label study of GSK2816126, an enhancer of zeste homolog 2 (EZH2) inhibitor, in patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL), transformed follicular lymphoma (tFL), other non-Hodgkin’s lymphomas (NHL), multiple myeloma (MM) and solid tumor. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Alzrigat, M.; Jernberg-Wiklund, H. The miR–125a and miR-320c are potential tumor suppressor microRNAs epigenetically silenced by the polycomb repressive complex 2 in multiple myeloma. RNA Dis. 2017, 4, e1529. [Google Scholar] [PubMed]
- Baughn, L.B.; di Liberto, M.; Niesvizky, R.; Cho, H.J.; Jayabalan, D.; Lane, J.; Liu, F.; Chen-Kiang, S. CDK2 phosphorylation of Smad2 disrupts TGF-beta transcriptional regulation in resistant primary bone marrow myeloma cells. J. Immunol. 2009, 182, 1810–1817. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ding, L.; Zhang, H.; Han, J.; Yang, X.; Yan, J.; Zhu, Y.; Li, J.; Song, H.; Ye, Q. Potentiation of Smad-mediated transcriptional activation by the RNA-binding protein RBPMS. Nucleic Acids Res. 2006, 34, 6314–6326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, A.C.; Vizán, P.; Hill, C.S. Smad3 protein levels are modulated by RAS activity and during the cell cycle to dictate transforming growth factor-beta responses. J. Biol. Chem. 2010, 285, 6489–6497. [Google Scholar] [CrossRef] [PubMed]
- Alzrigat, M.; Parraga, A.A.; Majumder, M.M.; Ma, A.; Jin, J.; Osterborg, A.; Nahi, H.; Nilsson, K.; Heckman, C.A.; Oberg, F.; et al. The polycomb group protein BMI-1 inhibitor PTC-209 is a potent anti–myeloma agent alone or in combination with epigenetic inhibitors targeting EZH2 and the BET bromodomains. Oncotarget 2017, 8, 103731–103743. [Google Scholar] [CrossRef] [PubMed]
- Hemming, S.; Cakouros, D.; Vandyke, K.; Davis, M.J.; Zannettino, A.C.; Gronthos, S. Identification of novel EZH2 targets regulating osteogenic differentiation in mesenchymal stem cells. Stem Cells Dev. 2016, 25, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Hemming, S.; Cakouros, D.; Isenmann, S.; Cooper, L.; Menicanin, D.; Zannettino, A.; Gronthos, S. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells 2014, 32, 802–815. [Google Scholar] [CrossRef] [PubMed]
- D'Souza, S.; del Prete, D.; Jin, S.; Sun, Q.; Huston, A.J.; Kostov, F.E.; Sammut, B.; Hong, C.S.; Anderson, J.L.; Patrene, K.D.; et al. Gfi1 expressed in bone marrow stromal cells is a novel osteoblast suppressor in patients with multiple myeloma bone disease. Blood 2011, 118, 6871–6880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamik, J.; Jin, S.; Sun, Q.; Zhang, P.; Weiss, K.R.; Anderson, J.L.; Silbermann, R.; Roodman, G.D.; Galson, D.L. EZH2 or HDAC1 inhibition reverses multiple myeloma–induced epigenetic suppression of osteoblast differentiation. Mol. Cancer Res. 2017, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| EZH2 Inhibitor | Anti-MM Activity | Treatment Type | Reference |
|---|---|---|---|
| UNC1999 | In vitro and in vivo | Single agent treatment or in combination with Bortezomib | [102,112,113] |
| GSK343 | In vitro | Single agent treatment | [58,102,107] |
| GSK126 | In vitro and in vivo | Single agent treatment or in combination with Bortezomib and Panobinostat | [107,114,115] |
| EPZ-7438 | In vitro and in vivo | Single agent treatment or in combination with Lenalidomide, Pomalidomide, Bortezomib and Panobinostat | [115,116,117] |
| EPZ005687 | In vitro | Single agent treatment | [88] |
| OR-S1 and OR-S2 | In vitro | Single agent treatment | [118] |
| GSK2816126 | Phase I clinical trial (NCT02082977)–Terminated | Single agent treatment | [119] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzrigat, M.; Jernberg-Wiklund, H.; Licht, J.D. Targeting EZH2 in Multiple Myeloma—Multifaceted Anti-Tumor Activity. Epigenomes 2018, 2, 16. https://doi.org/10.3390/epigenomes2030016
Alzrigat M, Jernberg-Wiklund H, Licht JD. Targeting EZH2 in Multiple Myeloma—Multifaceted Anti-Tumor Activity. Epigenomes. 2018; 2(3):16. https://doi.org/10.3390/epigenomes2030016
Chicago/Turabian StyleAlzrigat, Mohammad, Helena Jernberg-Wiklund, and Jonathan D. Licht. 2018. "Targeting EZH2 in Multiple Myeloma—Multifaceted Anti-Tumor Activity" Epigenomes 2, no. 3: 16. https://doi.org/10.3390/epigenomes2030016
APA StyleAlzrigat, M., Jernberg-Wiklund, H., & Licht, J. D. (2018). Targeting EZH2 in Multiple Myeloma—Multifaceted Anti-Tumor Activity. Epigenomes, 2(3), 16. https://doi.org/10.3390/epigenomes2030016