Targeting HDAC Complexes in Asthma and COPD



Abstract
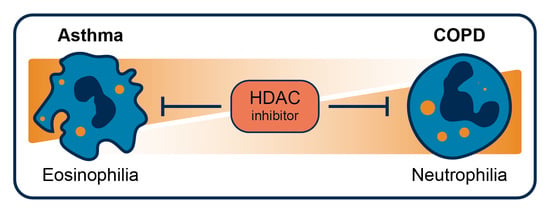
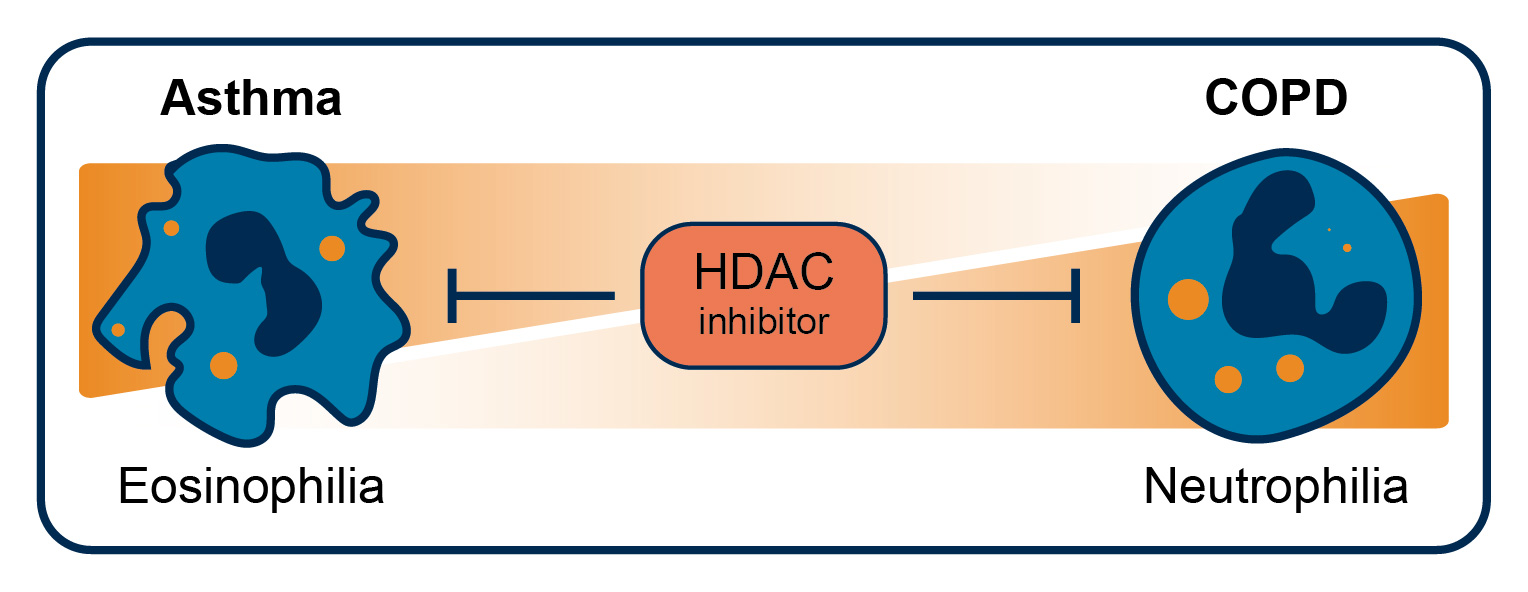
:
1. Introduction
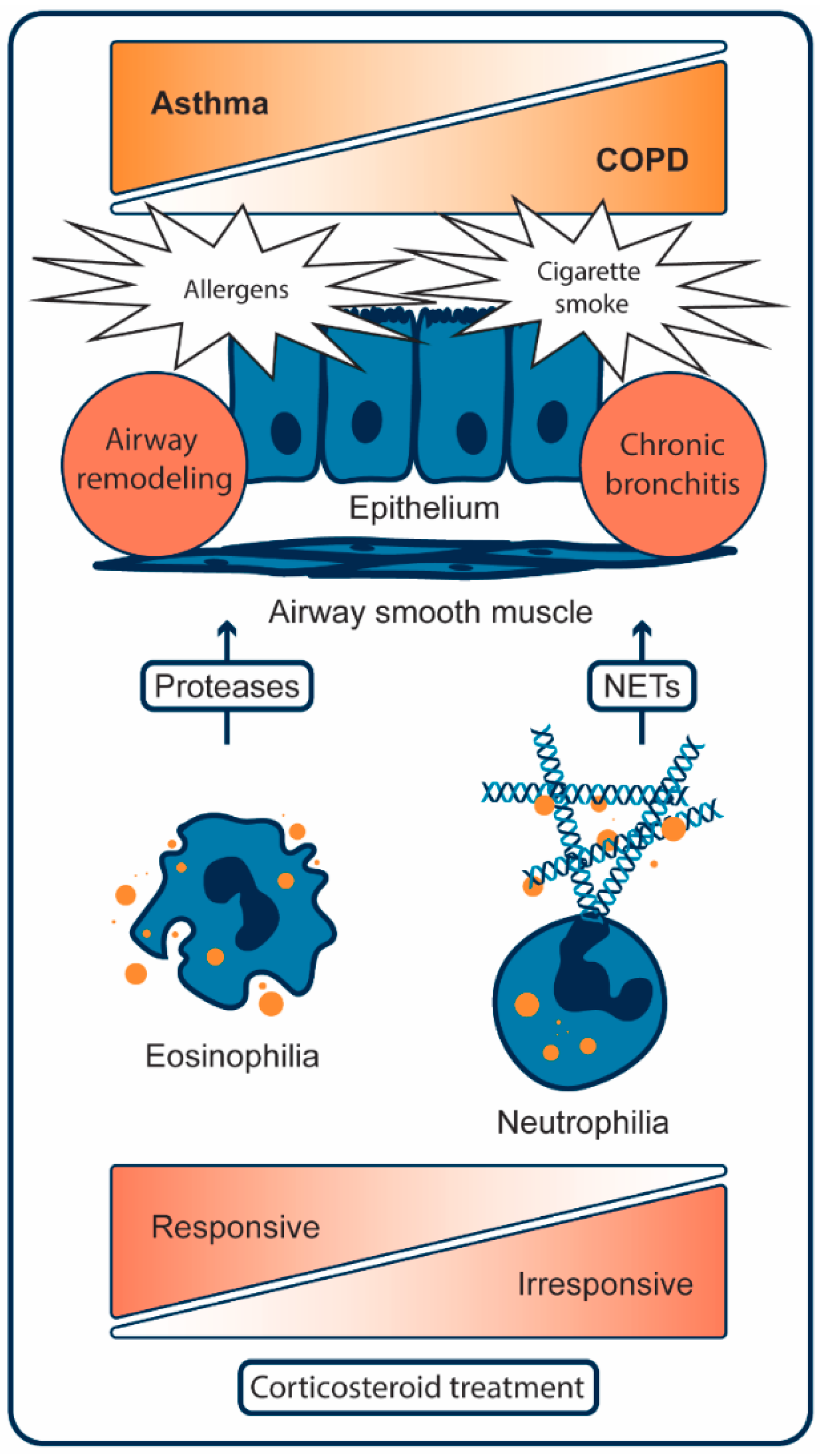
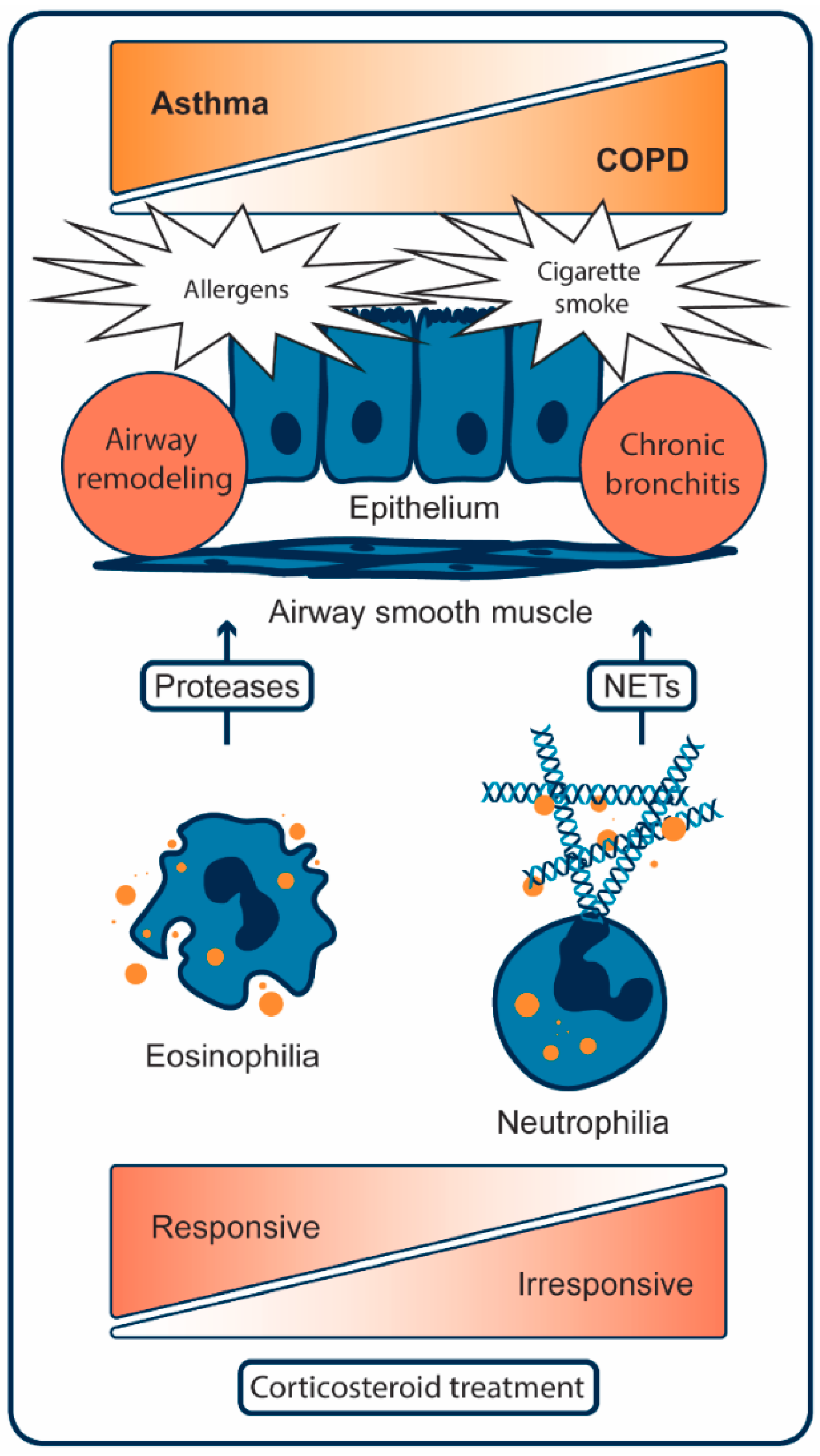
2. Pathogenesis of Asthma
2.1. Cellular Mechanisms of Asthma
2.2. Role of HDACs in Asthma
3. Pathogenesis of COPD
3.1. Cellular Mechanisms of COPD
3.2. Role of HDACs in COPD
4. The Role of NF-κB Acetylation in Asthma and COPD
5. HDACi in Asthma and COPD
6. Design of Selective HDACi Targeting the Catalytic Site
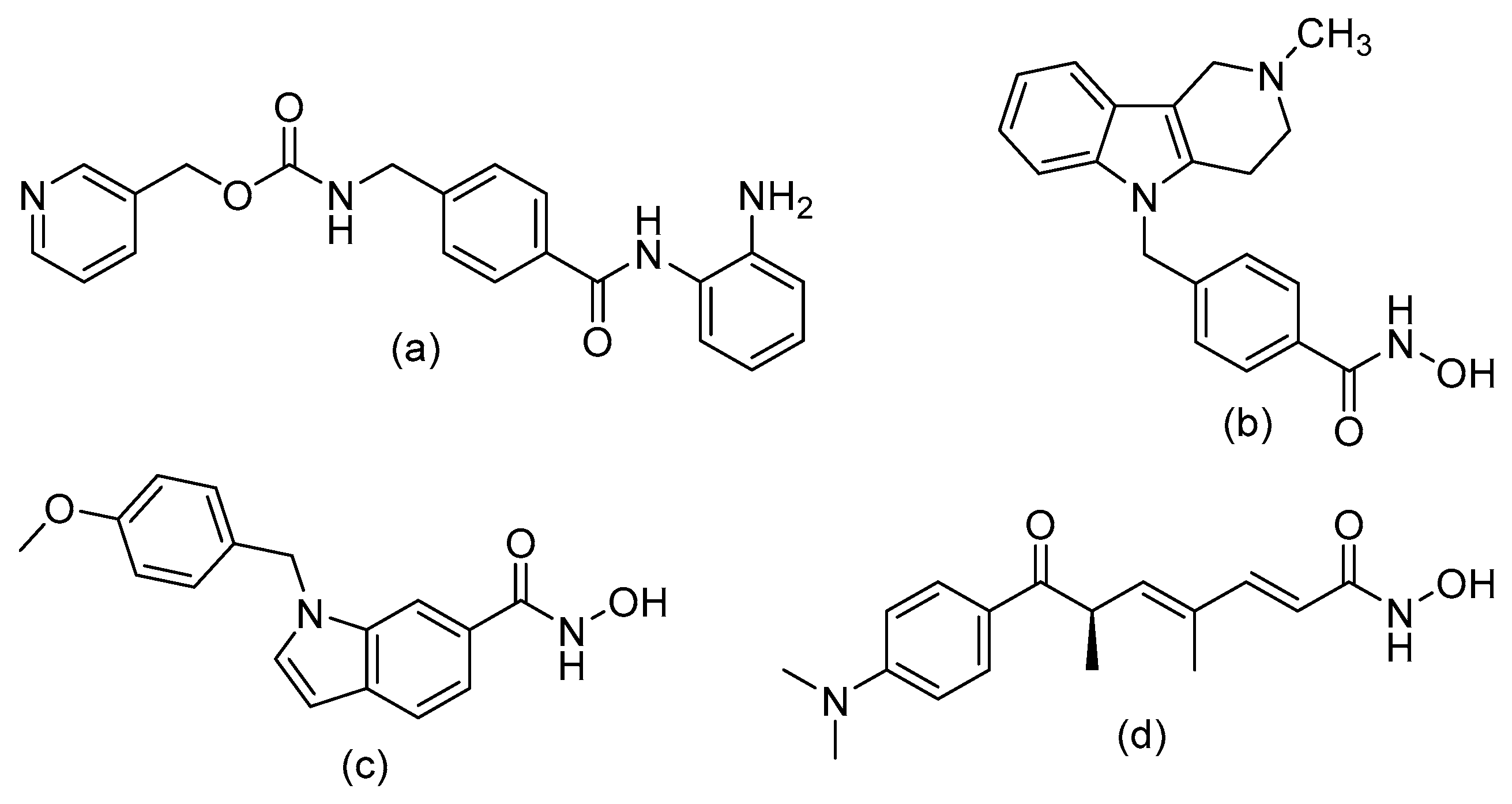
Structure–Activity Relationship of Reported o-Aminoanilides for Selective Inhibition of HDAC1/2/3
7. Targeting HDAC Complexes
7.1. Class I HDAC Complexes
7.2. The Sin3 Complex
7.2.1. Structure of Sin3
7.2.2. Roles of Sin3
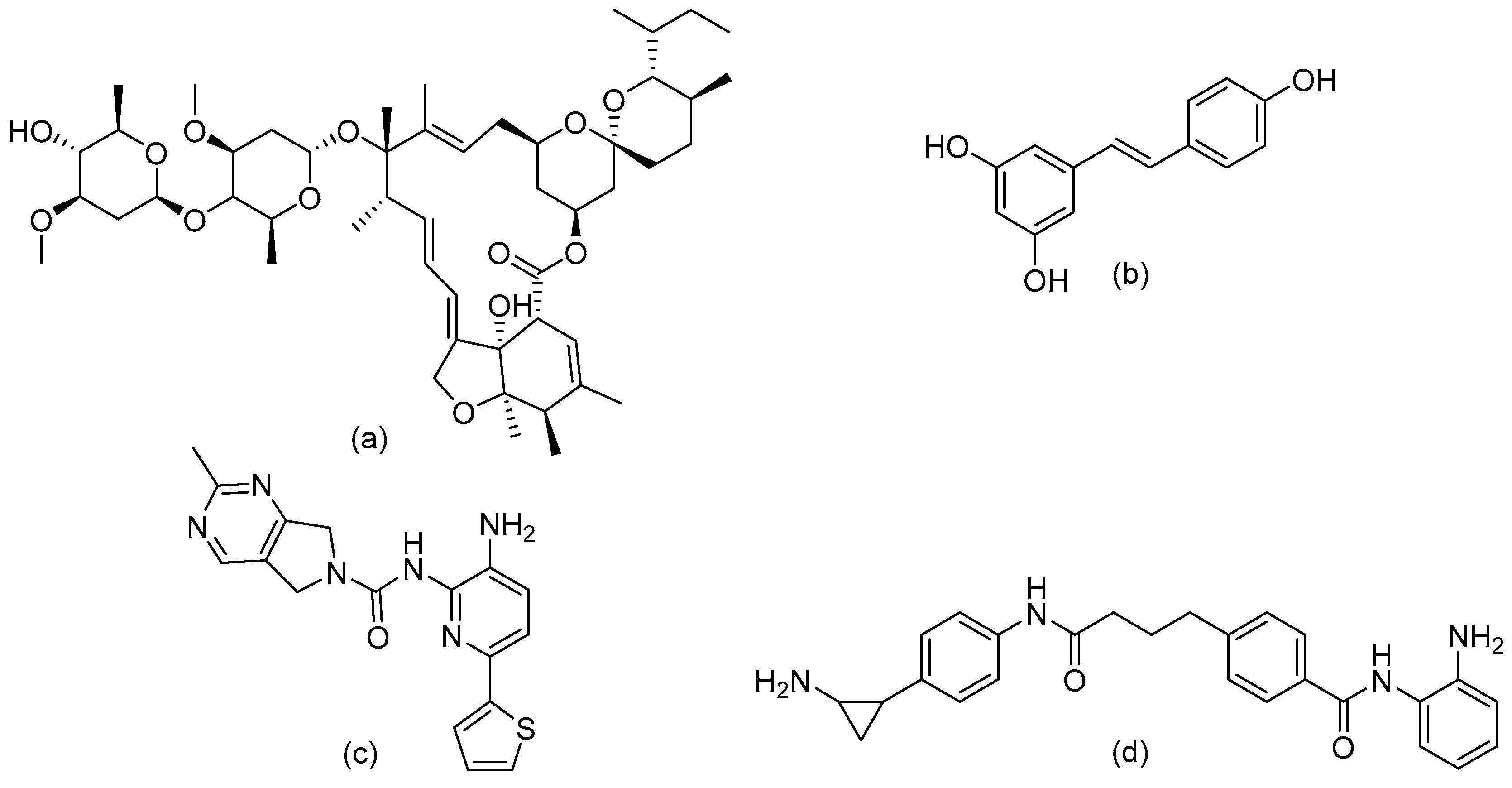
7.2.3. Examples of Sin3 Targeting
7.3. The NuRD Complex.
7.3.1. Structure of the NuRD Complex
7.3.2. Roles of NuRD
7.3.3. Targeting NuRD
7.4. The CoREST Complex
7.4.1. Structure and Roles of the CoREST Complex
7.4.2. Targeting CoREST
7.5. The SMRT/NCoR Complex
7.5.1. Structure of SMRT/NCoR
7.5.2. Roles of SMRT/NCoR
7.5.3. Targeting SMRT/NCoR
7.6. The Role of Inositol Phosphates in HDAC Complex Formation
8. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Addo-Yobo, E.O.; Ade, S.; Agodokpessi, G.; Aguirre, V.; Aït-Khaled, N. The Global Asthma Report 2018; The Global Asthma Network: Auckland, New Zealand, 2018. [Google Scholar]
- Croisant, S. Epidemiology of asthma: Prevalence and burden of disease. Adv. Exp. Med. Biol. 2014, 795, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Adeloye, D.; Chua, S.; Lee, C.; Basquill, C.; Papana, A.; Theodoratou, E.; Nair, H.; Gasevic, D.; Sridhar, D.; Campbell, H.; et al. Global Health Epidemiology Reference Group (GHERG) Global and regional estimates of COPD prevalence: Systematic review and meta-analysis. J. Glob. Health 2015, 5, 020415. [Google Scholar] [CrossRef] [PubMed]
- Lõpez-Campos, J.L.; Tan, W.; Soriano, J.B. Global burden of COPD. Respirology 2016, 21, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Leus, N.G.J.; Zwinderman, M.R.H.; Dekker, F.J. Histone deacetylase 3 (HDAC 3) as emerging drug target in NF-κB-mediated inflammation. Curr. Opin. Chem. Biol. 2016, 33, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.; Cusack, R.P.; Chaudhary, N.; Satia, I.; Kurmi, O.P. Under- and over-diagnosis of COPD: A global perspective. Breathe 2019, 15, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Wacker, M.E.; Jörres, R.A.; Karch, A.; Wilke, S.; Heinrich, J.; Karrasch, S.; Koch, A.; Schulz, H.; Watz, H.; Leidl, R.; et al. Assessing health-related quality of life in COPD: Comparing generic and disease-specific instruments with focus on comorbidities. BMC Pulm. Med. 2016, 16, 70. [Google Scholar] [CrossRef]
- Barnes, P.J.; Ito, K.; Adcock, I.M. Corticosteroid resistance in chronic obstructive pulmonary disease: Inactivation of histone deacetylase. Lancet 2004, 363, 731–733. [Google Scholar] [CrossRef]
- Becker, P.B.; Workman, J.L. Nucleosome remodeling and epigenetics. Cold Spring Harb. Perspect. Biol. 2013, 5, a017905. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Holgate, S.T. Pathogenesis of Asthma. Clin. Exp. Allergy 2008, 38, 872–897. [Google Scholar] [CrossRef]
- Fahy, J.V. Type 2 inflammation in asthma-present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Silkoff, P.E.; Moore, W.C.; Sterk, P.J. Three Major Efforts to Phenotype Asthma: Severe Asthma Research Program, Asthma Disease Endotyping for Personalized Therapeutics, and Unbiased Biomarkers for the Prediction of Respiratory Disease Outcome. Clin. Chest Med. 2019, 40, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Howarth, P.H.; Durham, S.R.; Lee, T.H.; Kay, A.B.; Church, M.K.; Holgate, S.T. Influence of albuterol, cromolyn sodium and ipratropium bromide on the airway and circulating mediator reponses to allergen bronchial provocation in asthma. Am. Rev. Respir. Dis. 1985, 132, 986–992. [Google Scholar] [PubMed]
- Cushley, M.; Tattersfield, A.; Holgate, S. Inhaled adenosine and guanosine on airway resistance in normal and asthmatic subjects. Br. J. Clin. Pharm. 2004, 58, S751–S755. [Google Scholar] [CrossRef] [PubMed]
- Bradding, P. Interleukin 4 is localized to and released by human mast cells. J. Exp. Med. 2004, 176, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Gour, N.; Wills-Karp, M. IL-4 and IL-13 signaling in allergic airway disease. Cytokine 2015, 75, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinke, J.W.; Borish, L. Th2 cytokines and asthma. Interleukin-4: Its role in the pathogenesis of asthma, and targeting it for asthma treatment with interleukin-4 receptor antagonists. Respir. Res. 2001, 2, 66–70. [Google Scholar] [CrossRef]
- Papathanassiou, E.; Loukides, S.; Bakakos, P. Severe asthma: Anti-IgE or anti-IL-5? Eur. Clin. Respir. J. 2016, 3, 31813. [Google Scholar] [CrossRef]
- Duvall, M.G.; Krishnamoorthy, N.; Levy, B.D. Non-type 2 inflammation in severe asthma is propelled by neutrophil cytoplasts and maintained by defective resolution. Allergol. Int. 2019, 68, 143–149. [Google Scholar] [CrossRef]
- Carr, T.F.; Zeki, A.A.; Kraft, M. Eosinophilic and noneosinophilic asthma. Am. J. Respir. Crit. Care Med. 2018, 197, 22–37. [Google Scholar] [CrossRef]
- Uddin, M.; Nong, G.; Ward, J.; Seumois, G.; Prince, L.R.; Wilson, S.J.; Cornelius, V.; Dent, G.; Djukanović, R. Prosurvival activity for airway neutrophils in severe asthma. Thorax 2010, 65, 684–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.; Watz, H.; Malmgren, A.; Pedersen, F. NETopathic inflammation in chronic obstructive pulmonary disease and severe asthma. Front. Immunol. 2019, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, F.; Waschki, B.; Marwitz, S.; Goldmann, T.; Kirsten, A.; Malmgren, A.; Rabe, K.F.; Uddin, M.; Watz, H. Neutrophil extracellular trap formation is regulated by CXCR2 in COPD neutrophils. Eur. Respir. J. 2018, 51, 1700970. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, N.; Douda, D.N.; Brüggemann, T.R.; Ricklefs, I.; Duvall, M.G.; Abdulnour, R.E.E.; Martinod, K.; Tavares, L.; Wang, X.; Cernadas, M.; et al. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci. Immunol. 2018, 3, eaao4747. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.E.; Chan, K. Interleukin-17 stimulates the expression of interleukin-8, growth-related oncogene-α, and granulocyte-colony-stimulating factor by human airway epithelial cells. Am. J. Respir. Cell Mol Biol. 2002, 26, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Al-Muhsen, S.; Johnson, J.R.; Hamid, Q. Remodeling in asthma. J. Allergy Clin. Immunol. 2011, 128, 451–462. [Google Scholar] [CrossRef]
- Uddin, M.; Lau, L.C.; Seumois, G.; Vijayanand, P.; Staples, K.J.; Bagmane, D.; Cornelius, V.; Dorinsky, P.; Davies, D.E.; Djukanović, R. EGF-induced bronchial epithelial cells drive neutrophil chemotactic and anti-apoptotic activity in asthma. PLoS ONE 2013, 8, e72502. [Google Scholar] [CrossRef]
- Ordoñez, C.L.; Khashayar, R.; Wong, H.H.; Ferrando, R.; Wu, R.; Hyde, D.M.; Hotchkiss, J.A.; Zhang, Y.; Novikov, A.; Dolganov, G.; et al. Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am. J. Respir. Crit. Care Med. 2001, 163, 517–523. [Google Scholar] [CrossRef]
- Blank, M.F.; Grummt, I. The seven faces of SIRT7. Transcription 2017, 8, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Altucci, L.; Rots, M.G. Epigenetic drugs: From chemistry via biology to medicine and back. Clin. Epigenet. 2016, 8, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone Deacetylase Inhibitors: Overview and Perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Zwinderman, M.R.H.; Dekker, F.J. The Process and Strategy for Developing Selective Histone Deacetylase 3 Inhibitors. Molecules 2018, 23, 551. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sahakian, E.; Powers, J.; Lienlaf, M.; Perez-Villarroel, P.; Knox, T.; Villagra, A. Functional analysis of histone deacetylase 11 (HDAC11). In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1436. [Google Scholar] [CrossRef]
- Su, R.C.; Becker, A.B.; Kozyrskyj, A.L.; HayGlass, K.T. Epigenetic regulation of established human type 1 versus type 2 cytokine responses. J. Allergy Clin. Immunol. 2008, 121, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Caramori, G.; Lim, S.; Oates, T.; Fan Chung, K.; Barnes, P.J.; Adcock, I.M. Expression and activity of histone deacetylases in human asthmatic airways. Am. J. Respir. Crit. Care Med. 2002, 166, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Seumois, G.; Chavez, L.; Gerasimova, A.; Lienhard, M.; Omran, N.; Kalinke, L.; Vedanayagam, M.; Ganesan, A.P.V.; Chawla, A.; Djukanović, R.; et al. Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility. Nat. Immunol. 2014, 15, 777–788. [Google Scholar] [CrossRef]
- Cosío, B.G.; Mann, B.; Ito, K.; Jazrawi, E.; Barnes, P.J.; Chung, K.F.; Adcock, I.M. Histone Acetylase and Deacetylase Activity in Alveolar Macrophages and Blood Mononocytes in Asthma. Am. J. Respir. Crit. Care Med. 2004, 170, 141–147. [Google Scholar] [CrossRef]
- Brook, P.O.; Perry, M.M.; Adcock, I.M.; Durham, A.L. Epigenome-modifying tools in asthma. Epigenomics 2015, 7, 1017–1032. [Google Scholar] [CrossRef] [Green Version]
- Butler, C.A.; McQuaid, S.; Taggart, C.C.; Weldon, S.; Carter, R.; Skibinski, G.; Warke, T.J.; Choy, D.F.; McGarvey, L.P.; Bradding, P.; et al. Glucocorticoid receptor β and histone deacetylase 1 and 2 expression in the airways of severe asthma. Thorax 2012, 67, 392–398. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, S.H.; Kim, Y.K.; Hong, S.J.; Min, K.U.; Cho, S.H.; Park, H.W. A polymorphism in the histone deacetylase 1 gene is associated with the response to corticosteroids in asthmatics. Korean J. Intern. Med. 2013, 28, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Petrache, I. Pathogenesis of chronic obstructive pulmonary disease. J. Clin. Investig. 2012, 122, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.D.; Goldstein, N.M.; Houghton, A.M.G.; Kobayashi, D.K.; Kelley, D.; Belaaouaj, A. Neutrophil Elastase Contributes to Cigarette Smoke-Induced Emphysema in Mice. Am. J. Pathol. 2003, 163, 2329–2335. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 2008, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Role of HDAC2 in the Pathophysiology of COPD. Annu. Rev. Physiol. 2009, 71, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Yang, Y.; Ma, D.; He, L.; Xiao, W. Expression level of histone deacetylase 2 correlates with occurring of chronic obstructive pulmonary diseases. Mol. Biol. Rep. 2013, 40, 3995–4000. [Google Scholar] [CrossRef]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.M.; Hogg, J.C.; et al. Decreased Histone Deacetylase Activity in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Rajendrasozhan, S.; Yang, S.-R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 177, 861–870. [Google Scholar] [CrossRef]
- Marwick, J.A.; Kirkham, P.A.; Stevenson, C.S.; Danahay, H.; Giddings, J.; Butler, K.; Donaldson, K.; MacNee, W.; Rahman, I. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am. J. Respir. Cell Mol. Biol. 2004, 31, 633–642. [Google Scholar] [CrossRef]
- Raij, L.; DeMaster, E.G.; Jaimes, E.A. Cigarette smoke-induced endothelium dysfunction: Role of superoxide anion. J. Hypertens. 2001, 19, 891–897. [Google Scholar] [CrossRef]
- Osoata, G.O.; Yamamura, S.; Ito, M.; Vuppusetty, C.; Adcock, I.M.; Barnes, P.J.; Ito, K. Nitration of distinct tyrosine residues causes inactivation of histone deacetylase 2. Biochem. Biophys. Res. Commun. 2009, 384, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Adenuga, D.; Yao, H.; March, T.H.; Seagrave, J.; Rahman, I. Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2009, 40, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Targeting the Epigenome in the Treatment of Asthma and Chronic Obstructive Pulmonary Disease. Proc. Am. Thorac. Soc. 2009, 6, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.A.S.; McCalman, M.T.; Moulos, P.; Francoijs, K.J.; Chatziioannou, A.; Kolisis, F.N.; Alexis, M.N.; Mitsiou, D.J.; Stunnenberg, H.G. Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res. 2011, 21, 1404–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Barnes, P.J.; Adcock, I.M. Glucocorticoid Receptor Recruitment of Histone Deacetylase 2 Inhibits Interleukin-1beta -Induced Histone H4 Acetylation on Lysines 8 and 12. Mol. Cell Biol. 2002, 20, 6891–6903. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2–mediated deacetylation of the glucocorticoid receptor enables NF-κB suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Van Der Velden, V.H.J. Glucocorticoids: Mechanisms of action and anti-inflammatory potential in asthma. Mediat. Inflamm. 1998, 7, 229–237. [Google Scholar] [CrossRef]
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645. [Google Scholar] [CrossRef]
- Mercado, N.; To, Y.; Ito, K.; Barnes, P.J. Nortriptyline Reverses Corticosteroid Insensitivity by Inhibition of Phosphoinositide-3-Kinase-. J. Pharm. Exp. 2011, 337, 465–470. [Google Scholar] [CrossRef]
- Ito, K.; Lim, S.; Caramori, G.; Cosio, B.; Chung, K.F.; Adcock, I.M.; Barnes, P.J. A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 8921–8926. [Google Scholar] [CrossRef] [Green Version]
- To, Y.; Ito, K.; Kizawa, Y.; Failla, M.; Ito, M.; Kusama, T.; Elliott, W.M.; Hogg, J.C.; Adcock, I.M.; Barnes, P.J. Targeting phosphoinositide-3-kinase-δ with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, D.H.; Lavender, P.; Seo, J.H.; Kim, Y.S.; Park, J.S.; Kwak, S.J.; Jee, Y.K. Repression of TNF-α-induced IL-8 expression by the glucocorticoid receptor-β involves inhibition of histone H4 acetylation. Exp. Mol. Med. 2009, 41, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.C.; Oakley, R.H.; Jewell, C.M.; Cidlowski, J.A. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative isoform: A mechanism for the generation of glucocorticoid resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 6865–6870. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Tello, A.; Semlali, A.; Chakir, J.; Martin, J.G.; Leung, D.Y.; Eidelman, D.H.; Hamid, Q. Induction of glucocorticoid receptor-β expression in epithelial cells of asthmatic airways by T-helper type 17 cytokines. Clin. Exp. Allergy 2010, 40, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Chen, X.; Liu, J.; bo Zhou, D.; Kuang, X.; Xiao, J.; Yu, Q.; Lu, X.; Li, W.; Xie, B.; et al. Serum IL-1β and IL-17 levels in patients with COPD: Associations with clinical parameters. Int. J. COPD 2017, 12, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Leung, D.Y.M.; Martin, R.J.; Goleva, E. Inhibition of Histone Deacetylase 2 Expression by Elevated Glucocorticoid Receptor β in Steroid-resistant Asthma. Am. J. Respir. Crit. Care Med. 2010, 182, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.C.; Chen, L.-F. Regulation of NF-κB Action by Reversible Acetylation. Reversible Protein Acetylation: Novartis Foundation Symposium; Wiley: Chichester, UK, 2004; Volume 259. [Google Scholar] [CrossRef]
- Kiernan, R.; Brès, V.; Ng, R.W.M.; Coudart, M.P.; El Messaoudi, S.; Sardet, C.; Jin, D.Y.; Emiliani, S.; Benkirane, M. Post-activation turn-off of NF-κB-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 2003, 278, 2758–2766. [Google Scholar] [CrossRef]
- Ziesché, E.; Kettner-Buhrow, D.; Weber, A.; Wittwer, T.; Jurida, L.; Soelch, J.; Müller, H.; Newel, D.; Kronich, P.; Schneider, H.; et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-κB. Nucleic Acids Res. 2013, 41, 90–109. [Google Scholar] [CrossRef]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef]
- Mujtaba, S.; Zeng, L.; Zhou, M.M. Structure and acetyl-lysine recognition of the bromodomain. Oncogene 2007, 26, 5521–5527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothgiesser, K.M.; Fey, M.; Hottiger, M.O. Acetylation of p65 at lysine 314 is important for late NF-κB-dependent gene expression. BMC Genom. 2010, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, X.-D.; Zhou, M.-M.; Ozato, K.; Chen, L.-F. Brd4 Coactivates Transcriptional Activation of NF- B via Specific Binding to Acetylated RelA. Mol. Cell Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Dekker, F.J.; Van Den Bosch, T.; Martin, N.I. Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov. Today 2014, 19, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Fossati, G.; Mascagni, P. Histone Deacetylase Inhibitors for Treating a Spectrum of Diseases Not Related to Cancer. Mol. Med. 2011, 17, 333–352. [Google Scholar] [CrossRef] [PubMed]
- Leus, N.G.J.; Van Der Wouden, P.E.; Van Den Bosch, T.; Hooghiemstra, W.T.R.; Ourailidou, M.E.; Kistemaker, L.E.M.; Bischoff, R.; Gosens, R.; Haisma, H.J.; Dekker, F.J. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-κB p65 transcriptional activity. Biochem. Pharm. 2016, 108, 58–74. [Google Scholar] [CrossRef]
- Leus, N.G.J.; Van Den Bosch, T.; Van Der Wouden, P.E.; Krist, K.; Ourailidou, M.E.; Eleftheriadis, N.; Kistemaker, L.E.M.; Bos, S.; Gjaltema, R.A.F.; Mekonnen, S.A.; et al. HDAC1-3 inhibitor MS-275 enhances IL10 expression in RAW264.7 macrophages and reduces cigarette smoke-induced airway inflammation in mice. Sci. Rep. 2017, 7, 1–18. [Google Scholar] [CrossRef]
- Waltregny, D.; Glénisson, W.; Tran, S.L.; North, B.J.; Verdin, E.; Colige, A.; Castronovo, V. Histone deacetylase HDAC8 associates with smooth muscle α-actin and is essential for smooth muscle cell contractility. FASEB J. 2005, 19, 966–968. [Google Scholar] [CrossRef]
- Ren, Y.; Su, X.; Kong, L.; Li, M.; Zhao, X.; Yu, N.; Kang, J. Therapeutic effects of histone deacetylase inhibitors in a murine asthma model. Inflamm. Res. 2016, 65, 995–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.; Xie, S.; Liu, Y.; Liu, W.; Li, D.; Liu, M.; Luo, H.R.; Zhou, J. Histone deacetylase 6 modulates macrophage infiltration during inflammation. Theranostics 2018, 8, 2927–2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, A.D.-A.; Marmo, T.P.; Salam, A.A.; Che, S.; Finkelstein, E.; Kabarriti, R.; Xenias, H.S.; Mazitschek, R.; Hubbert, C.; Kawaguchi, Y.; et al. HDAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. J. Cell Sci. 2007, 120, 1469–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Haspel, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K.; et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Investig. 2013, 123, 5212–5230. [Google Scholar] [CrossRef] [PubMed]
- Toki, S.; Goleniewska, K.; Reiss, S.; Zhou, W.; Newcomb, D.C.; Bloodworth, M.H.; Stier, M.T.; Boyd, K.L.; Polosukhin, V.V.; Subramaniam, S.; et al. The histone deacetylase inhibitor trichostatin A suppresses murine innate allergic inflammation by blocking group 2 innate lymphoid cell (ILC2) activation. Thorax 2016, 71, 633–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Oh, S.W.; Kang, M.S.; Kwon, H.J.; Oh, G.T.; Kim, D.Y. Trichostatin A attenuates airway inflammation in mouse asthma model. Clin. Exp. Allergy 2005, 35, 89–96. [Google Scholar] [CrossRef]
- Kankaanranta, H.; Janka-Junttila, M.; Ilmarinen-Salo, P.; Ito, K.; Jalonen, U.; Ito, M.; Adcock, I.M.; Moilanen, E.; Zhang, X. Histone deacetylase inhibitors induce apoptosis in human eosinophils and neutrophils. J. Inflamm. 2010, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Hamam, H.J.; Palaniyar, N. Histone Deacetylase Inhibitors Dose-Dependently Switch Neutrophil Death from NETosis to Apoptosis. Biomolecules 2019, 9, 184. [Google Scholar] [CrossRef]
- Grabiec, A.M.; Krausz, S.; de Jager, W.; Burakowski, T.; Groot, D.; Sanders, M.E.; Prakken, B.J.; Maslinski, W.; Eldering, E.; Tak, P.P.; et al. Histone Deacetylase Inhibitors Suppress Inflammatory Activation of Rheumatoid Arthritis Patient Synovial Macrophages and Tissue. J. Immunol. 2010, 184, 2718–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabiec, A.M.; Hussell, T. The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin. Immunopathol. 2016, 38, 409–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, I.K.H.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Penberthy, K.K.; Juncadella, I.J.; Ravichandran, K.S. Apoptosis and engulfment by bronchial epithelial cells: Implications for allergic airway inflammation. Ann. Am. Thorac. Soc. 2014, 11, S259–S262. [Google Scholar] [CrossRef] [PubMed]
- Mayo, M.W.; Denlinger, C.E.; Broad, R.M.; Yeung, F.; Reilly, E.T.; Shi, Y.; Jones, D.R. Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF-κB through the Akt pathway. J. Biol. Chem. 2003, 278, 18980–18989. [Google Scholar] [CrossRef]
- Marek, M.; Shaik, T.B.; Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Ramos-Morales, E.; Da Veiga, C.; Kalinin, D.; Melesina, J.; Robaa, D.; et al. Characterization of Histone Deacetylase 8 (HDAC8) Selective Inhibition Reveals Specific Active Site Structural and Functional Determinants. J. Med. Chem. 2018, 61, 10000–10016. [Google Scholar] [CrossRef] [PubMed]
- Wambua, M.K.; Nalawansha, D.A.; Negmeldin, A.T.; Pflum, M.K.H. Mutagenesis studies of the 14 Å internal cavity of histone deacetylase 1: Insights toward the acetate-escape hypothesis and selective inhibitor design. J. Med. Chem. 2014, 57, 642–650. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Chen, Y.P.P. Ligand release mechanisms and channels in histone deacetylases. J. Comput. Chem. 2013, 34, 2270–2283. [Google Scholar] [CrossRef]
- Suzuki, T.; Ando, T.; Tsuchiya, K.; Fukazawa, N.; Saito, A.; Mariko, Y.; Yamashita, T.; Nakanishi, O. Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J. Med. Chem. 1999, 42, 3001–3003. [Google Scholar] [CrossRef]
- Wagner, F.F.; Weïwer, M.; Steinbacher, S.; Schomburg, A.; Reinemer, P.; Gale, J.P.; Campbell, A.J.; Fisher, S.L.; Zhao, W.N.; Reis, S.A.; et al. Kinetic and structural insights into the binding of histone deacetylase 1 and 2 (HDAC1, 2) inhibitors. Bioorganic Med. Chem. 2016, 24, 4008–4015. [Google Scholar] [CrossRef]
- Methot, J.L.; Chakravarty, P.K.; Chenard, M.; Close, J.; Cruz, J.C.; Dahlberg, W.K.; Fleming, J.; Hamblett, C.L.; Hamill, J.E.; Harrington, P.; et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorganic Med. Chem. Lett. 2008, 18, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.Y.; Chuang, H.C.; Shen, F.H.; Detroja, K.; Hsin, L.W.; Chen, C.S. Targeting breast cancer stem cells by novel HDAC3-selective inhibitors. Eur. J. Med. Chem. 2017, 140, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.F.; Lundh, M.; Kaya, T.; McCarren, P.; Zhang, Y.L.; Chattopadhyay, S.; Gale, J.P.; Galbo, T.; Fisher, S.L.; Meier, B.C.; et al. An Isochemogenic Set of Inhibitors to Define the Therapeutic Potential of Histone Deacetylases in β-Cell Protection. ACS Chem. Biol. 2016, 11, 363–374. [Google Scholar] [CrossRef] [PubMed]
- McClure, J.J.; Inks, E.S.; Zhang, C.; Peterson, Y.K.; Li, J.; Chundru, K.; Lee, B.; Buchanan, A.; Miao, S.; Chou, C.J. Comparison of the Deacylase and Deacetylase Activity of Zinc-Dependent HDACs. ACS Chem. Biol. 2017, 12, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Sasaki, T.; Kanki, H.; Choong, C.J.; Nishiyama, K.; Kubo, G.; Hotei, A.; Taniguchi, M.; Mochizuki, H.; Uesato, S. New 5-Aryl-Substituted 2-Aminobenzamide-Type HDAC Inhibitors with a Diketopiperazine Group and Their Ameliorating Effects on Ischemia-Induced Neuronal Cell Death. Sci. Rep. 2018, 8, 1400. [Google Scholar] [CrossRef] [PubMed]
- Witter, D.J.; Harrington, P.; Wilson, K.J.; Chenard, M.; Fleming, J.C.; Haines, B.; Kral, A.M.; Secrist, J.P.; Miller, T.A. Optimization of biaryl Selective HDAC1&2 Inhibitors (SHI-1:2). Bioorganic Med. Chem. Lett. 2008, 18, 726–731. [Google Scholar] [CrossRef]
- Ayer, D.E. Histone deacetylases: Transcriptional repression with SINers and NuRDs. Trends Cell Biol. 1999, 9, 193–198. [Google Scholar] [CrossRef]
- Khan, D.H.; He, S.; Yu, J.; Winter, S.; Cao, W.; Seiser, C.; Davie, J.R. Protein kinase CK2 regulates the dimerization of histone deacetylase 1 (HDAC1) and HDAC2 during mitosis. J. Biol. Chem. 2013, 288, 16518–16528. [Google Scholar] [CrossRef] [PubMed]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Targeting class I histone deacetylases in cancer therapy. Expert Opin. Ther. Targets 2013, 17, 29–41. [Google Scholar] [CrossRef]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef]
- Becher, I.; Dittmann, A.; Savitski, M.M.; Hopf, C.; Drewes, G.; Bantscheff, M. Chemoproteomics reveals time-dependent binding of histone deacetylase inhibitors to endogenous repressor complexes. ACS Chem. Biol. 2014, 9, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharm. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.A.; Ekwall, K. Sin3: A flexible regulator of global gene expression and genome stability. Curr. Genet. 2005, 47, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Alland, L.; Muhle, R.; Hou, H.; Potes, J.; Chin, L.; Schreiber-Agus, N.; DePinho, R.A. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature 1997, 387, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Kadamb, R.; Mittal, S.; Bansal, N.; Batra, H.; Saluja, D. Sin3: Insight into its transcription regulatory functions. Eur. J. Cell Biol. 2013, 92, 237–246. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Tong, J.K.; Schreiber, S.L. Genomewide studies of histone deacetylase function in yeast. Proc. Natl. Acad. Sci. USA 2000, 97, 13708–13713. [Google Scholar] [CrossRef] [Green Version]
- Dannenberg, J.H.; David, G.; Zhong, S.; Van Der Torre, J.; Wong, W.H.; DePinho, R.A. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005, 19, 1581–1595. [Google Scholar] [CrossRef] [Green Version]
- Pile, L.A.; Spellman, P.T.; Katzenberger, R.J.; Wassarman, D.A. The SIN3 Deacetylase Complex Represses Genes Encoding Mitochondrial Proteins. J. Biol. Chem. 2003, 278, 37840–37848. [Google Scholar] [CrossRef] [Green Version]
- Saha, N.; Liu, M.; Gajan, A.; Pile, L.A. Genome-wide studies reveal novel and distinct biological pathways regulated by SIN3 isoforms. BMC Genom. 2016, 17, 111. [Google Scholar] [CrossRef]
- Chaubal, A.; Pile, L.A. Same agent, different messages: Insight into transcriptional regulation by SIN3 isoforms. Epigenet. Chromatin. 2018, 11, 17. [Google Scholar] [CrossRef]
- Bainor, A.J.; Saini, S.; Calderon, A.; Casado-Polanco, R.; Giner-Ramirez, B.; Moncada, C.; Cantor, D.J.; Ernlund, A.; Litovchick, L.; David, G. The HDAC-Associated Sin3B Protein Represses DREAM Complex Targets and Cooperates with APC/C to Promote Quiescence. Cell Rep. 2018, 25, 2797–2807. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Sangodkar, J.; Negre, N.; Narla, G.; Cagan, R.L. Sin3a acts through a multi-gene module to regulate invasion in Drosophila and human tumors. Oncogene 2013, 32, 3184–3197. [Google Scholar] [CrossRef] [PubMed]
- Cantor, D.J.; David, G. The potential of targeting sin3b and its associated complexes for cancer therapy. Expert Opin. Ther. Targets 2017, 21, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.-J.; Petrie, K.; Leibovitch, B.A.; Zeng, L.; Mezei, M.; Howell, L.; Gil, V.; Christova, R.; Bansal, N.; Yang, S.; et al. Selective Inhibition of SIN3 Corepressor with Avermectins as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2015, 14, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Ci, X.; Chen, N.; Chen, C.; Li, X.; Chu, X.; Li, J.; Deng, X. Anti-inflammatory effects of ivermectin in mouse model of allergic asthma. Inflamm. Res. 2011, 60, 589–596. [Google Scholar] [CrossRef]
- Laing, R.; Gillan, V.; Devaney, E. Ivermectin – Old Drug, New Tricks? Trends Parasitol. 2017, 33, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Bird, A.; Reinberg, D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999, 13, 1924–1935. [Google Scholar] [CrossRef]
- Gnanapragasam, M.N.; Scarsdale, J.N.; Amaya, M.L.; Webb, H.D.; Desai, M.A.; Walavalkar, N.M.; Wang, S.Z.; Zu Zhu, S.; Ginder, G.D.; Williams, D.C. p66 -MBD2 coiled-coil interaction and recruitment of Mi-2 are critical for globin gene silencing by the MBD2-NuRD complex. Proc. Natl. Acad. Sci. USA 2011, 108, 7487–7492. [Google Scholar] [CrossRef]
- Xue, Y.; Wong, J.; Moreno, G.T.; Young, M.K.; Côté, J.; Wang, W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell 1998, 2, 851–861. [Google Scholar] [CrossRef]
- Millard, C.J.; Watson, P.J.; Celardo, I.; Gordiyenko, Y.; Cowley, S.M.; Robinson, C.V.; Fairall, L.; Schwabe, J.W.R. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol. Cell 2013, 51, 57–67. [Google Scholar] [CrossRef]
- Millard, C.J.; Varma, N.; Saleh, A.; Morris, K.; Watson, P.J.; Bottrill, A.R.; Fairall, L.; Smith, C.J.; Schwabe, J.W. The structure of the core NuRD repression complex provides insights into its interaction with chromatin. Elife 2016, 5, e13941. [Google Scholar] [CrossRef] [PubMed]
- Schmidberger, J.W.; Sharifi Tabar, M.; Torrado, M.; Silva, A.P.G.; Landsberg, M.J.; Brillault, L.; AlQarni, S.; Zeng, Y.C.; Parker, B.L.; Low, J.K.K.; et al. The MTA1 subunit of the nucleosome remodeling and deacetylase complex can recruit two copies of RBBP4/7. Protein Sci. 2016, 25, 1472–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basta, J.; Rauchman, M. The Nucleosome Remodeling and Deacetylase Complex in Development and Disease. In Translating Epigenetics to the Clinic; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Williams, C.J.; Naito, T.; Gómez-Del Arco, P.; Seavitt, J.R.; Cashman, S.M.; De Souza, B.; Qi, X.; Keables, P.; Von Andrian, U.H.; Georgopoulos, K. The chromatin remodeler Mi-2β is required for CD4 expression and T cell development. Immunity 2004, 20, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Stephens, L.C.; Kumar, R. Metastasis tumor antigen family proteins during breast cancer progression and metastasis in a reliable mouse model for human breast cancer. Clin. Cancer Res. 2006, 12, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Wang, R.A.; Bagheri-Yarmand, R. Emerging Roles of MTA Family Members in Human Cancers. In Seminars in Oncology; Elsevier: Amsterdam, The Netherlands, 2003; Volume 30. [Google Scholar] [CrossRef]
- Pakala, S.B.; Bui-Nguyen, T.M.; Reddy, S.D.N.; Li, D.Q.; Peng, S.; Rayala, S.K.; Behringer, R.R.; Kumar, R. Regulation of NF-κB circuitry by a component of the nucleosome remodeling and deacetylase complex controls inflammatory response homeostasis. J. Biol. Chem. 2010, 285, 23590–23597. [Google Scholar] [CrossRef]
- Dhar, S.; Kumar, A.; Zhang, L.; Rimando, A.M.; Lage, J.M.; Lewin, J.R.; Atfi, A.; Zhang, X.; Levenson, A.S. Dietary pterostilbene is a novel MTA1-targeted chemopreventive and therapeutic agent in prostate cancer. Oncotarget 2016, 7, 18469–18484. [Google Scholar] [CrossRef] [PubMed]
- Kai, L.; Samuel, S.K.; Levenson, A.S. Resveratrol enhances p53 acetylation and apoptosis in prostate cancer by inhibiting MTA1/NuRD complex. Int. J. Cancer 2010, 126, 1538–1548. [Google Scholar] [CrossRef]
- Butt, N.A.; Kumar, A.; Dhar, S.; Rimando, A.M.; Akhtar, I.; Hancock, J.C.; Lage, J.M.; Pound, C.R.; Lewin, J.R.; Gomez, C.R.; et al. Targeting MTA1/HIF-1α signaling by pterostilbene in combination with histone deacetylase inhibitor attenuates prostate cancer progression. Cancer Med. 2017, 6, 2673–2685. [Google Scholar] [CrossRef]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2002, 98, 1454–1458. [Google Scholar] [CrossRef]
- Andres, M.E.; Burger, C.; Peral-Rubio, M.J.; Battaglioli, E.; Anderson, M.E.; Grimes, J.; Dallman, J.; Ballas, N.; Mandel, G. CoREST: A functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. USA 2002, 96, 9873–9878. [Google Scholar] [CrossRef]
- Barrios, Á.P.; Gómez, A.V.; Sáez, J.E.; Ciossani, G.; Toffolo, E.; Battaglioli, E.; Mattevi, A.; Andrés, M.E. Differential properties of transcriptional complexes formed by the CoREST family. Mol. Cell Biol. 2014, 34, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Gocke, C.B.; Luo, X.; Borek, D.; Tomchick, D.R.; Machius, M.; Otwinowski, Z.; Yu, H. Structural Basis for CoREST-Dependent Demethylation of Nucleosomes by the Human LSD1 Histone Demethylase. Mol. Cell 2006, 23, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Abrajano, J.J.; Qureshi, I.A.; Gokhan, S.; Zheng, D.; Bergman, A.; Mehler, M.F. REST and CoREST modulate neuronal subtype specification, maturation and maintenance. PLoS ONE 2009, 4, e7936. [Google Scholar] [CrossRef] [PubMed]
- Fuller, N.O.; Pirone, A.; Lynch, B.A.; Hewitt, M.C.; Quinton, M.S.; McKee, T.D.; Ivarsson, M. CoREST Complex-Selective Histone Deacetylase Inhibitors Show Prosynaptic Effects and an Improved Safety Profile to Enable Treatment of Synaptopathies. ACS Chem. Neurosci. 2019, 10, 1729–1743. [Google Scholar] [CrossRef]
- Kalin, J.H.; Wu, M.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.; Panova, I.; Chung, H.J.; et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 2018, 9, 53. [Google Scholar] [CrossRef]
- Hörlein, A.J.; Näär, A.M.; Heinzel, T.; Torchia, J.; Gloss, B.; Kurokawa, R.; Ryan, A.; Kamei, Y.; Söderström, M.; Glass, C.K.; et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 1995, 377, 397–404. [Google Scholar] [CrossRef]
- Chen, J.D.; Evans, R.M. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 1995, 377, 454–457. [Google Scholar] [CrossRef]
- Guenther, M.G.; Barak, O.; Lazar, M.A. The SMRT and N-CoR Corepressors Are Activating Cofactors for Histone Deacetylase 3. Mol. Cell Biol. 2002, 21, 6091–6101. [Google Scholar] [CrossRef]
- Guenther, M.G.; Lane, W.S.; Fischle, W.; Verdin, E.; Lazar, M.A.; Shiekhattar, R. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 2000, 14, 1048–1057. [Google Scholar]
- Zhang, J.; Kalkum, M.; Chait, B.T.; Roeder, R.G. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell 2002, 9, 611–623. [Google Scholar] [CrossRef]
- Oberoi, J.; Fairall, L.; Watson, P.J.; Yang, J.C.; Czimmerer, Z.; Kampmann, T.; Goult, B.T.; Greenwood, J.A.; Gooch, J.T.; Kallenberger, B.C.; et al. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat. Struct. Mol. Biol. 2011, 18, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.M.; Watson, P.J.; Fairall, L.; Jamieson, A.G.; Schwabe, J.W.R. Insights into the recruitment of class IIa histone deacetylases (HDACs) to the SMRT/NCoR transcriptional repression complex. J. Biol. Chem. 2015, 290, 18237–18244. [Google Scholar] [CrossRef]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfi, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef] [Green Version]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef]
- Heinzel, T.; Lavinsky, R.M.; Mullen, T.M.; Söderström, M.; Laherty, C.D.; Torchia, J.; Yang, W.M.; Brard, G.; Ngo, S.D.; Davie, J.R.; et al. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature 1997, 387, 43–48. [Google Scholar] [CrossRef]
- Hermanson, O.; Jepsen, K.; Rosenfeld, M.G. N-CoR controls differentiation of neural stem cells into astrocytes. Nature 2002, 419, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, K.; Solum, D.; Zhou, T.; McEvilly, R.J.; Kim, H.J.; Glass, C.K.; Hermanson, O.; Rosenfeld, M.G. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 2007, 450, 415–419. [Google Scholar] [CrossRef]
- Jepsen, K.; Gleiberman, A.S.; Shi, C.; Simon, D.I.; Rosenfeld, M.G. Cooperative regulation in development by SMRT and FOXP1. Genes Dev. 2008, 22, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Mullican, S.E.; Gaddis, C.A.; Alenghat, T.; Nair, M.G.; Giacomin, P.R.; Everett, L.J.; Feng, D.; Steger, D.J.; Schug, J.; Artis, D.; et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011, 25, 2480–2488. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, S.; Lemmens, S.; Baeten, P.; Sommer, D.; Dooley, D.; Hendrix, S.; Gou Fabregas, M. HDAC3 Inhibition Promotes Alternative Activation of Macrophages but Does Not Affect Functional Recovery after Spinal Cord Injury. Exp. Neurobiol. 2018, 27, 437. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Perissi, V.; Aggarwal, A.; Glass, C.K.; Rose, D.W.; Rosenfeld, M.G. A Corepressor/Coactivator Exchange Complex Required for Transcriptional Activation by Nuclear Receptors and Other Regulated Transcription Factors. Cell 2004, 116, 511–526. [Google Scholar] [CrossRef] [Green Version]
- Ghisletti, S.; Huang, W.; Jepsen, K.; Benner, C.; Hardiman, G.; Rosenfeld, M.G.; Glass, C.K. Cooperative NCoR/SMRT interactions establish a eorepressor-based strategy for integration of inflammatory ana anti-inflammatory signaling pathways. Genes Dev. 2009, 23, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Jamaladdin, S.; Kelly, R.D.W.; O’Regan, L.; Dovey, O.M.; Hodson, G.E.; Millard, C.J.; Portolano, N.; Fry, A.M.; Schwabe, J.W.R.; Cowley, S.M. Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 9840–9845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worley, J.; Luo, X.; Capaldi, A.P. Inositol Pyrophosphates Regulate Cell Growth and the Environmental Stress Response by Activating the HDAC Rpd3L. Cell Rep. 2013, 3, 1476–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W.R. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012, 481, 335–340. [Google Scholar] [CrossRef]
- Watson, P.J.; Millard, C.J.; Riley, A.M.; Robertson, N.S.; Wright, L.C.; Godage, H.Y.; Cowley, S.M.; Jamieson, A.G.; Potter, B.V.L.; Schwabe, J.W.R. Insights into the activation mechanism of class i HDAC complexes by inositol phosphates. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Arrar, M.; Turnham, R.; Pierce, L.; De Oliveira, C.A.F.; Andrew McCammon, J. Structural insight into the separate roles of inositol tetraphosphate and deacetylase-activating domain in activation of histone deacetylase 3. Protein Sci. 2013, 22, 83–92. [Google Scholar] [CrossRef]
- Angiolilli, C.; Kabala, P.A.; Grabiec, A.M.; Van Baarsen, I.M.; Ferguson, B.S.; García, S.; Fernandez, B.M.; McKinsey, T.A.; Tak, P.P.; Fossati, G.; et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann. Rheum. Dis. 2017, 76, 277–285. [Google Scholar] [CrossRef]
- Olson, D.E.; Wagner, F.F.; Kaya, T.; Gale, J.P.; Aidoud, N.; Davoine, E.L.; Lazzaro, F.; Weïwer, M.; Zhang, Y.L.; Holson, E.B. Discovery of the first histone deacetylase 6/8 dual inhibitors. J. Med. Chem. 2013, 56, 4816–4820. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorganic Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Lv, W.; Su, S.; Wu, W.; Rao, Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toure, M.; Crews, C.M. Small-molecule PROTACS: New approaches to protein degradation. Angew. Chem. Int. Ed. 2016, 55, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87. [Google Scholar] [CrossRef] [PubMed]
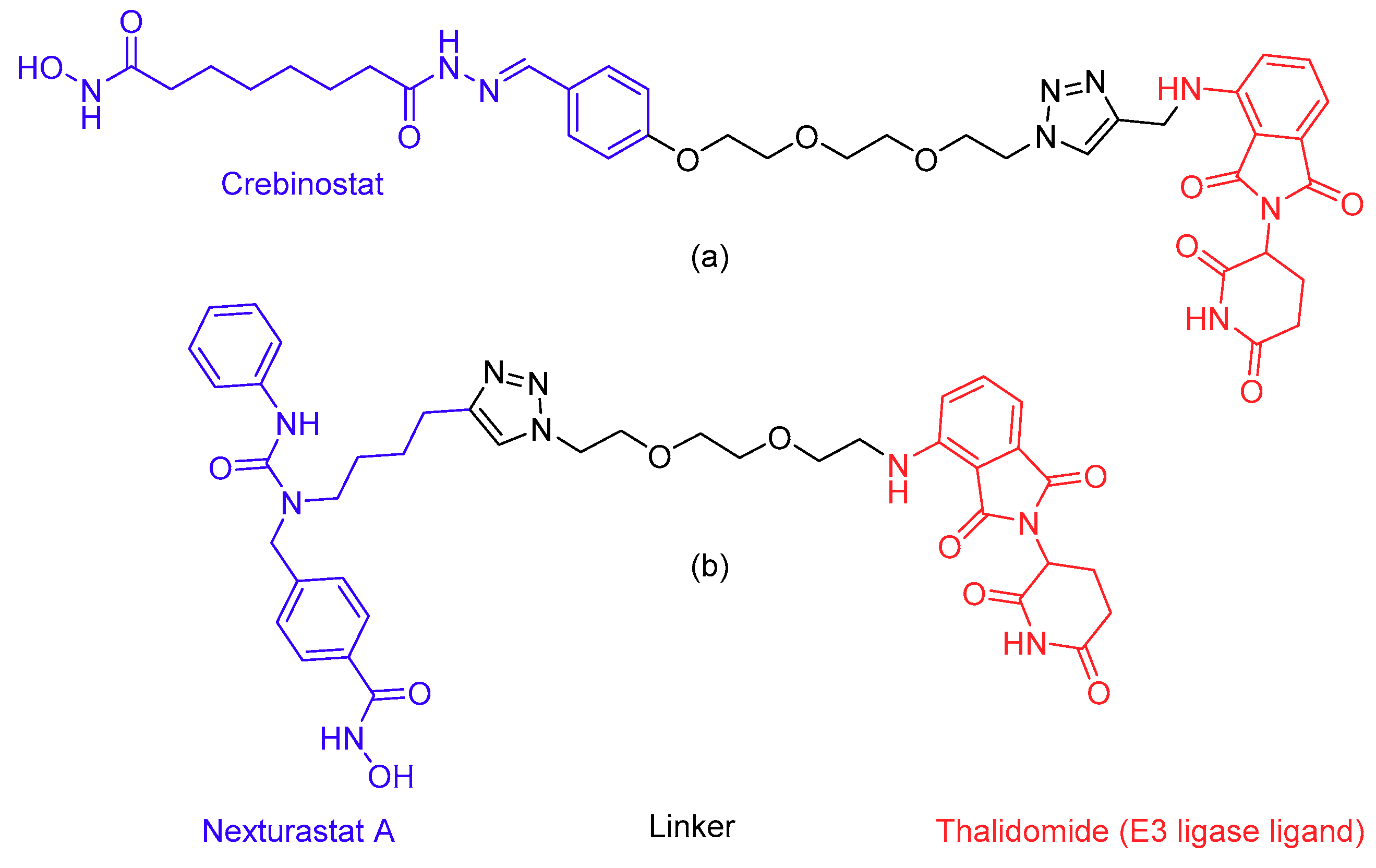
- Fass, D.M.; Reis, S.A.; Ghosh, B.; Hennig, K.M.; Joseph, N.F.; Zhao, W.N.; Nieland, T.J.F.; Guan, J.S.; Groves Kuhnle, C.E.; Tang, W.; et al. Crebinostat: A novel cognitive enhancer that inhibits histone deacetylase activity and modulates chromatin-mediated neuroplasticity. Neuropharmacology 2013, 64, 81–96. [Google Scholar] [CrossRef]
- Bergman, J.A.; Woan, K.; Perez-Villarroel, P.; Villagra, A.; Sotomayor, E.M.; Kozikowski, A.P. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J. Med. Chem. 2012, 55, 9891–9899. [Google Scholar] [CrossRef]


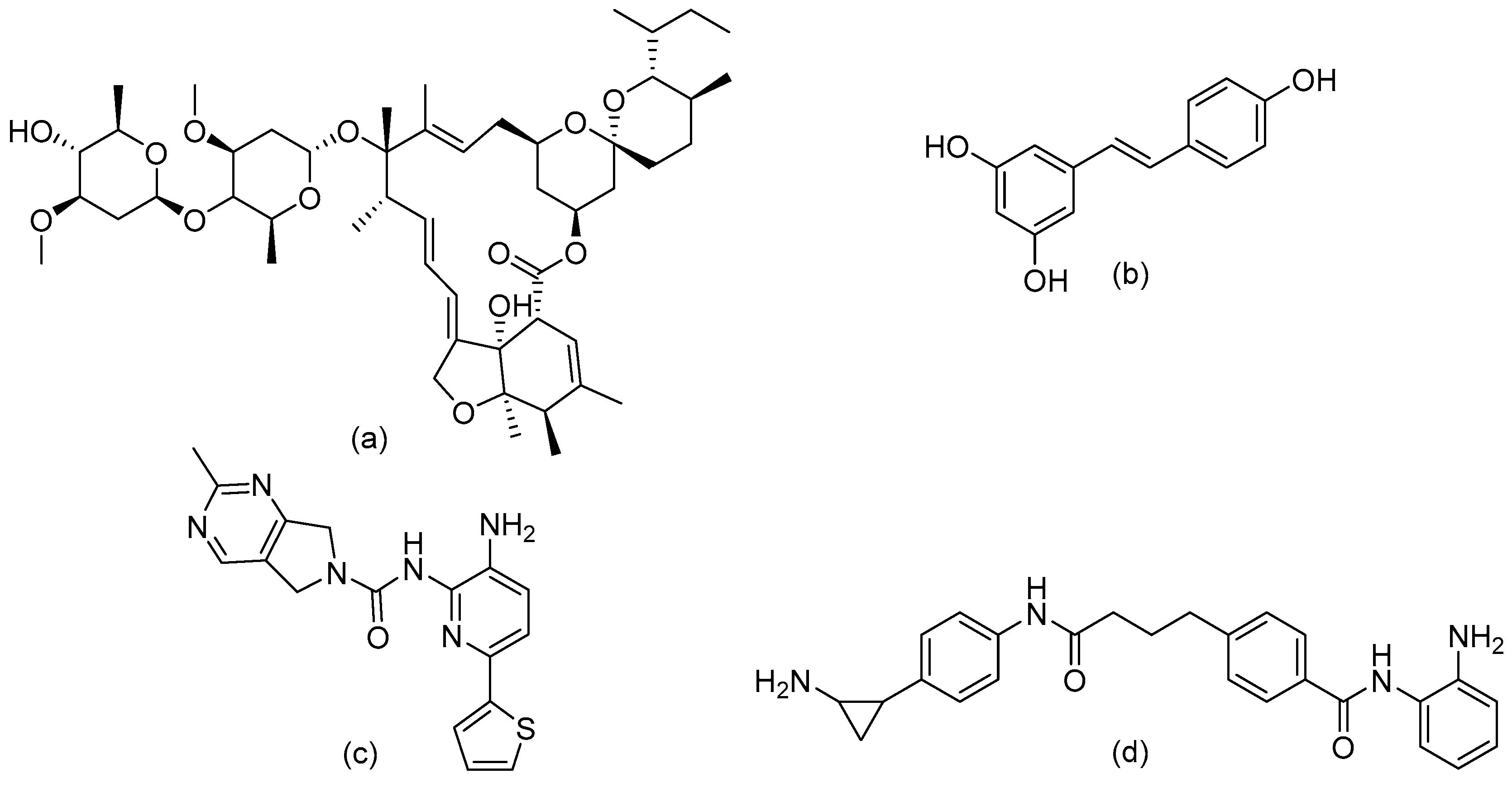


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In Vivo Model | Histone Deacetylase | Effect | ||||||
|---|---|---|---|---|---|---|---|---|
| Inhibitor | 1 | 2 | 3 | 6 | 8 | |||
| Asthma | Chronic asthmatic mouse model [84] | Tubastatin A | − | − | − | + | − | Reduced inflammation |
| PCI-34051 | − | − | − | − | + | Reduced hyperresponsiveness and inflammation | ||
| Murine innate allergic lung inflammation [90] | Trichostatin A | + | + | + | + | + | Decreased amount of inflammatory cells and inflammatory proteins | |
| Chronic Obstructive Pulmonary Disease | Cigarette smoke exposed mice [82] | Entinostat | + | + | + | − | − | Reduced expression of IL-8 and decreased influx of neutrophils |
| Cigarette smoke exposed mice [89] | Tubastatin A | − | − | − | + | − | Protection from cigarette-smoke induced mucociliary clearance disruption | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zwinderman, M.R.H.; de Weerd, S.; Dekker, F.J. Targeting HDAC Complexes in Asthma and COPD. Epigenomes 2019, 3, 19. https://doi.org/10.3390/epigenomes3030019
Zwinderman MRH, de Weerd S, Dekker FJ. Targeting HDAC Complexes in Asthma and COPD. Epigenomes. 2019; 3(3):19. https://doi.org/10.3390/epigenomes3030019
Chicago/Turabian StyleZwinderman, Martijn R. H., Sander de Weerd, and Frank J. Dekker. 2019. "Targeting HDAC Complexes in Asthma and COPD" Epigenomes 3, no. 3: 19. https://doi.org/10.3390/epigenomes3030019
APA StyleZwinderman, M. R. H., de Weerd, S., & Dekker, F. J. (2019). Targeting HDAC Complexes in Asthma and COPD. Epigenomes, 3(3), 19. https://doi.org/10.3390/epigenomes3030019