Abstract
Metformin, which is used as a first line treatment for type 2 diabetes mellitus (T2DM), has been shown to affect epigenetic patterns. In this study, we investigated the DNA methylation and potential lncRNA modifications in metformin-treated and newly diagnosed adults with T2DM. Genome-wide DNA methylation and lncRNA analysis were performed from the peripheral blood of 12 screen-detected and 12 metformin-treated T2DM individuals followed by gene ontology (GO) and KEGG pathway analysis. Differentially methylated regions (DMRs) observed showed 22 hypermethylated and 11 hypomethylated DMRs between individuals on metformin compared to screen-detected subjects. Amongst the hypomethylated DMR regions were the SLC gene family, specifically, SLC25A35 and SLC28A1. Fifty-seven lncRNA-associated DNA methylation regions included the mitochondrial ATP synthase-coupling factor 6 (ATP5J). Functional gene mapping and pathway analysis identified regions in the axon initial segment (AIS), node of Ranvier, cell periphery, cleavage furrow, cell surface furrow, and stress fiber. In conclusion, our study has identified a number of DMRs and lncRNA-associated DNA methylation regions in metformin-treated T2DM that are potential targets for therapeutic monitoring in patients with diabetes.
1. Introduction
DNA methylation, the most widely studied epigenetic mechanism, involves the covalent addition of a methyl group at the 5′ position of the cytosine ring within the 5′-CpG-3′ dinucleotides to create a 5-methylcytosine (5-mC). The target of DNA methylation, catalyzed by DNA methyltransferases (DNMTs) enzymes, are CpG nucleotides, which are usually unmethylated [1]. These CpG nucleotides occur at high-frequency in the promoter regions of genes and are frequently associated with hyper- or hypomethylation events [2]. Hypermethylation of promoter CpG islands can result in suppression of gene expression, whereas hypomethylation is associated with the transcriptional activation of affected genes [3]. Various studies suggest that these modifications may alter the transcriptional activity of genes and contribute to pathogenic conditions, such as the type 2 diabetes mellitus (T2DM) phenotype [4,5]. Studies also indicate that response to anti-diabetic agents and occurrence of diabetes complications can result from the actions of DNA methylation [4,6].
Although progression of disease cannot solely be attributed to DNA methylation, the impact of long non-coding RNAs (lncRNAs) on biological and pathologic processes have also been linked to various conditions including cancers and metabolic diseases [7,8]. LncRNAs are transcription products greater than 200 nucleotides with limited protein coding function [9]. They have been implicated in the regulation gene expression at the epigenetic, transcriptional, and post-transcription level [10]. Studies show that lncRNAs may also play a role in the diagnosis and therapeutic management of diabetes due to their involvement in regulatory processes and complications of T2DM [11,12].
Metformin, a drug commonly used for the treatment of T2DM, is highly effective with minimal side effects [13]. It has the ability to promote the phosphorylation and activation of AMP-activated protein kinase (AMPK), which results in the inhibition of gluconeogenic genes. In addition to glucose metabolism, the activation of AMPK impacts other pathways, such as lipid metabolism, mitochondrial biogenesis, autophagy, cell growth, and circadian rhythm [14]. Once activated, AMPK phosphorylates epigenetic enzymes, such as DNA methyltransferases (DNMTs), resulting in their inhibition [15]. The effects of metformin on DNA methylation include both hypo- and hypermethylation at the promoters of different genes, which in turn, could act to enhance or suppress gene expression [16,17,18,19]. Metformin was shown to affect DNA methylation even in healthy individuals immediately 10 h after drug administration [19]. These alterations in DNA methylation has also been evident in cancer related studies, showing that DNA methylation plays a role in the antidiabetic and potential anti-cancer actions of metformin [15,20,21,22].
Despite recent advances in the role of DNA methylation and diabetes, data on its effect in those under treatment with metformin in Africa are lacking. We, therefore, aimed to characterize the DNA methylation modifications in newly diagnosed and metformin-treated South Africans with T2DM. The knowledge gained could be used as a basis for further studies to elucidate the role of DNA methylation in the monitoring and treatment of T2DM within a South African context.
2. Results
2.1. Clinical Characteristics of the Study Population
The general clinical characteristics of the study population are summarized in Table 1. The study sample comprised 24 participants—12 screen-detected and 12 metformin-treated T2DM. There were no significant differences between the two groups in all the clinical characteristics. The duration of disease in the metformin-treated T2DM ranged from 0.5 to 17 with an average of 5.2 years.

Table 1.
Clinical characteristics of the study population.
2.2. Differentially Methylated Regions, LncRNA-Associated DNA Methylation, Gene Ontology (GO) and Pathway Analysis
A total of 33 differentially methylated regions (DMRs) were observed between individuals on metformin treatment compared to screen-detected subjects. Of these, 22 were hypermethylated, whilst another 11 were hypomethylated in participants treated with metformin, and these are summarized in Table 2. Lnc-associated DNA methylation peaks in the promoter regions are summarized in Table 3 showing that 36 were hypermethylated, and 21 were hypomethylated in individuals on metformin. KEGG pathway analysis revealed no enriched pathways. Based on GO analyses, we retrieved the biological process, cellular process, and molecular function of the DMRs, and these are presented in Figure 1 and Figure 2. The top enrichment scores for cellular processes of hypermethylated DMRs in subjects on metformin were associated with the axon initial segment, node of Ranvier, cell periphery, cleavage furrow, cell surface furrow, and stress fiber (Figure 1), whilst the hypomethylated biological processes were associated with photoreceptor outer segment (Figure 2).
Table 2.
Differentially methylated regions (DMRs) in T2DM on metformin versus newly diagnosed cases.
Table 3.
LncRNA-associated DNA methylation peaks of known diabetes versus screen-detected diabetes.
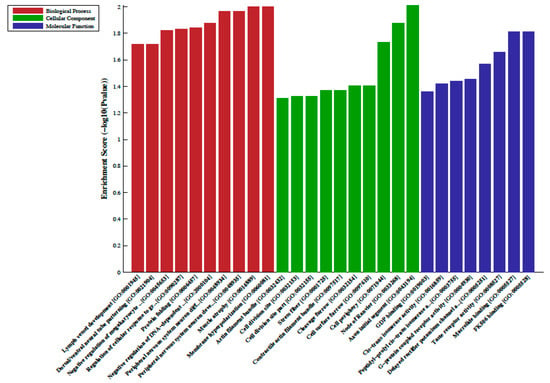
Figure 1.
Gene ontology (GO) enrichment analysis of the differentially hypermethylated genes in metformin-treated diabetes. The bar plot shows the top ten enrichment score values of the significant enrichment terms. Enriched GO terms were categorized into biological processes, cellular components, and molecular function. Data are presented as enriched scores expressed as −log10 (p value).
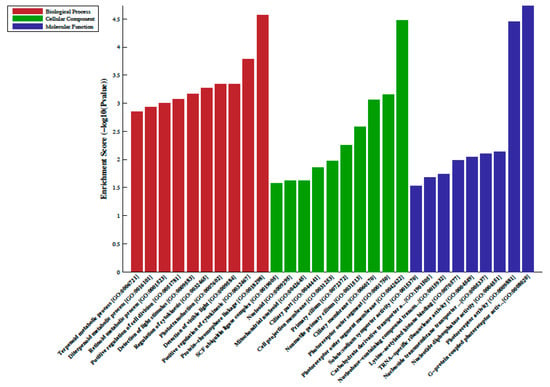
Figure 2.
Gene ontology (GO) enrichment analysis of the differentially hypomethylated genes in metformin-treated diabetes. The bar plot shows the top ten enrichment score values of the significant enrichment terms. Enriched GO terms were categorized into biological processes, cellular components, and molecular function. Data are presented as enriched scores expressed as −log10 (p value).
3. Discussion
In this study, we measured DNA methylation in diabetic individuals on metformin treatment compared to newly diagnosed diabetes cases and found 33 differentially methylated regions (DMRs) of which 22 (67%) were hypermethylated in diabetes subjects on metformin therapy. Furthermore, 57 lncRNA-associated DNA Methylation regions (36 hypermethylated and 21 hypomethylated) were detected of which 63% were hypermethylated in metformin-treated subjects. Functional pathway analysis of these DMRs revealed that they affect gene expression in the axon initial segment (AIS), node of Ranvier, cell periphery, cleavage furrow, cell surface furrow, and stress fiber.
Amongst the hypomethylated DMRs found in this study were genes in the SLC family, specifically SLC25A35 and SLC28A1. The SLC family is known for its importance in drug development, and their proteins include passive transporters, symporters, and antiporters and are located in cellular and organelle membranes [23]. Transporters facilitate the movement of a specific substrate across the membrane with or against its concentration gradient and sequence analysis of SLC25A35 indicates that it likely functions as an oxaloacetate carrier, implying mitochondrial association [24]. On the other hand, SLC28A1, a high-affinity pyrimidine nucleoside transporter, plays a role in renal reabsorption and has been observed to be impaired during diabetes [25]. Metformin treatment has been associated with lower methylation levels in SLC transporter genes, as was shown in a study conducted on metformin transporter genes in liver tissue [18]. Mitochondrial dysfunction due to diabetes affects oxidative phosphorylation and decreases ATP production. As SLC proteins transport various solutes across the mitochondrial membrane in order to partake in a number of metabolic pathways [26,27], the decrease in methylation and subsequent increase in gene expression of SLC transporters could be indicative of the antidiabetic effect of metformin treatment. It is, therefore, likely that metformin in its demethylation action of SLC mitochondrial carriers could possibly aid cell repair in these patients, however, this requires further investigation.
Functional pathway analysis observed in this study is consistent with the basic pathological abnormalities in Diabetic Peripheral Neuropathy (DPN), such as axonal degeneration and demyelination, lack of sensation, numbness, paresthesia, and allodynia experienced by diabetic individuals [28]. Cell death of nerves in DPN results from multifactorial metabolic imbalances associated with diabetes. The resulting mitochondrial dysfunction through a series of cascade effects involving AMP-activated protein kinase (AMPK), sirtuin (SIRT), and peroxisome proliferator-activated receptor-γ coactivator α (PGCα) suppresses mitochondrial oxidative phosphorylation, resulting in neuronal and axonal degeneration through increased oxidative injury [29,30].
Treatment with metformin was shown to decrease the incidence of DPN as was observed by the Bypass Angioplasty Revascularization Investigation 2 Diabetes trial [31]. Although metformin cannot reverse the nerve damage caused by diabetes, it could assist in managing blood glucose levels and improving the symptoms for patients.
In addition to DMRs, 57 lncRNA-associated DNA Methylation Peaks were detected when comparing known diabetic individuals to screen detected patients. Most recently the NONCODE database has updated the numbers of human lncRNAs to 167,150 with numbers still increasing [32]. Recent genome-wide association studies (GWAS) have shown positive correlation of some lncRNAs and diabetes [33]. In a related study, Sathishkumar et al. (2018) found increased levels of lncRNAs in T2DM patients, including HOTAIR, MEG3, LET, MALAT1, MIAT, CDKN2BAS1/ANRIL, XIST, PANDA, GAS5, Linc-p21, ENST00000550337.1, PLUTO, NBR2THRIL, and SALRNA1. The majority of these lncRNAs were involved in cell cycle regulation and senescence with their expression levels correlating to poor glycemic control, insulin resistance, and inflammation [11]. Similarly, HECTD4 and MBTPS1 were identified as the target genes for lncRNAs ENST00000364558 and ENST00000565382, respectively, with involvement in the development of T2DM by means of the lysosome and phagocytic signaling pathways [34]. Our findings indicate several novel lncRNA, including a lncRNA associated with the mitochondrial ATP synthase-coupling factor 6 (ATP5J) enzyme thought to be involved in the oxidative phosphorylation pathway [35]. Our data suggest higher methylation levels of this lncRNA in metformin-treated subjects, possibly pointing to suppression of this lncRNA allowing for ATP5J expression. Although little association was found between metformin and lncRNAs in our study, the significant novel lncRNA identified warrants further investigation to explore possible roles in type 2 diabetes.
The limitations of this study include the small sample size and the inclusion of women only; however, this allowed comparison and limited error that may result in statistical manipulation of a small sample size by sex. Furthermore, we used peripheral blood DNA to perform the genome-wide DNA methylation analysis. Epigenetic changes are believed to be organ specific; however, investigations on peripheral blood DNA have shown consistent methylation patterns with other organs [36,37]. Although the average (5.2 years) duration of disease in metformin-treated subjects was within the four to six years in which a person may have had the condition before clinical diagnosis [38], these findings should be interpreted with caution. In conclusion, our study has identified a number of DMRs and lncRNA-associated DNA methylation regions in metformin-treated T2DM that are potential targets for therapeutic monitoring in diabetes patients. However, these findings require further longitudinal study investigations that can clearly ascertain that these observations are not confounded by the duration and severity of diabetes.
4. Materials and Methods
4.1. Ethical Approval of the Study
This investigation used data from the Cape Town Vascular and Metabolic Health (VMH) study), which were approved by the Research Ethics Committees of the Cape Peninsula University of Technology and Stellenbosch University (resp., NHREC: REC-230 408-014 and N14/01/003; approved date: 21 May 2018). The Code of Ethics of the World Medical Association (Declaration of Helsinki) was also applied to the study. Signed written consent was obtained from all participants after all procedures were explained in the language of their choice.
4.2. Study Procedures
In this case-control study, the participants were females matched for both age and body mass index. All study participants underwent a standardized interview, blood pressure, and anthropometric measurements. A 75 g oral glucose tolerance test (OGTT) was performed on participants with no previous diagnosis of diabetes mellitus. Participants who met the World Health Organisation (WHO) criteria for diabetes were termed as screen-detected or newly diagnosed diabetes. Biochemical parameters analyzed at an ISO 15189 accredited pathology practice (PathCare, Reference Laboratory, Cape Town, South Africa) included the following: plasma glucose, serum insulin, serum creatinine, total cholesterol (TC), high-density lipoprotein cholesterol (HDL-c), triglycerides (TG), low-density lipoprotein cholesterol (LDL), C-reactive protein (CRP), γ-glutamyl transferase (GGT), AST, ALT, and glycated hemoglobin (HbA1c), certified by the National Glycohemoglobin Standardization Program (NGSP). In addition, a full blood count was also done for all participants, and ethylenediaminetetraacetic acid (EDTA) treated blood samples were stored at −20 degrees Celsius for DNA extraction and analysis.
4.3. Genome-Wide DNA Methylation Sequencing
Genomic DNA was extracted from peripheral blood using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. At least 2 µg of DNA (concentrations ranging between 70 and 130 ng/μL) with A260/A280 and A260/A230 ratios ≥ 1.8 was shipped frozen on dry ice, as instructed by Arraystar Inc. (Rockville, MD, USA). Methylated DNA immunoprecipitation (MeDIP) was performed by Arraystar Inc. (Rockville, MD, USA) according to Down et al. [39], with minor modifications as follows. About 1 μg of fragmented DNA was prepared for Illumina HiSeq 4000 sequencing as the following steps: (1) end repair of DNA samples with T4 DNA polymerase, Klenow DNA polymerase, and T4 PNK; (2) a single ‘A’ base was added to the 3’ ends with Klenow (exo minus) polymerase; (3) Illumina’s genomic adapters were ligated to DNA fragments; (4) DNA fragments were immunoprecipitated by anti-5-methylcytosine antibody (Diagenode); (5) immunoprecipitated DNA fragments were amplified by PCR amplification; (6) ~300–600 bp DNA fragments were extracted by gel purification. The completed libraries were quantified by Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The libraries were denatured with 0.1 M NaOH to generate single-stranded DNA molecules, captured on Illumina flow cell, amplified in situ. The libraries were then sequenced on the Illumina HiSeq 4000 following the TruSeq SBS Kit v5 protocol. The enrichment of DNA immunoprecipitation was analyzed by qPCR using specific methylated sites at H19 locus and non-methylated sites at GAPDH.
4.4. MeDIP-Seq Data Analysis
The enrichment of DNA immunoprecipitation was analyzed by qPCR using specific methylated sites at H19 locus and non-methylated sites at GAPDH. Image analysis and base calling were performed using Off-Line Basecaller software (OLB V1.8). After passing a Solexa CHASTITY quality filter, the clean reads were aligned to the human genome (UCSC HG19) using HISAT2 software (V2.1.0). Briefly, individual bases generated from original image files have quality scores, which reflect the probability whether base calling is correct or not. The score is calculated by CHASTITY Formula. The CHASTITY (C) of each base in the short reads is determined by the intensity of four colors (IA, IC, IG, and IT here), and the formula means “the ratio of the highest (IC here) of the four (base type) intensities to the sum of highest two (IC and IG here).” The CHASTITY (C) should be no less than 0.6 in the first 25 bases. Statistically significant MeDIP-enriched regions (peaks) detected by MACS v2 were identified by comparison to input background, using a q-value threshold of 10−5. The peaks in samples were annotated by the nearest gene using the newest UCSC RefSeq database. Differentially methylated regions (DMRs) located within gene promoters (TSS − 2000 bp, TSS + 2000 bp) with statistical significance between the two groups were identified by diffReps (Cut-off: log2FC = 1.0, p-value = 10−4).
4.5. Gene Ontology (GO) and KEGG Pathway Analysis
The ontology covers three domains, namely biological process, cellular component, and molecular function. Fisher’s exact test was used to determine whether there was more overlap between the DE list and the GO annotation list than would be expected by chance. The p value denotes the significance of GO terms enrichment in the DE genes. The lower the p value, the more significant the GO term; a p value ≤ 0.05 was considered significant. Annotation was performed using standard workflow according to http://geneontology.org/. Pathway analysis was done using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The p value (EASE score, Fisher’s p-value, or hypergeometric p-value) denotes the significance of the pathway correlated to the conditions. The lower the p value is, the more significant the pathway is; a p value ≤ 0.05 was considered significant.
Author Contributions
W.L.S.: wrote the first draft, experimental procedures, data analysis, and interpretation. S.B.E.H.: co-drafted the manuscript, statistical analysis, and interpretation of data. S.R.: interpretation of data, editing and revising it for intellectual content. R.T.E.: conception, interpretation of the data, revising it for intellectual content and final approval of the version to be published. A.P.K.: conception, interpretation of the data, revising it for intellectual content and final approval of the version to be published. T.E.M.: conception and design of the study, analysis and interpretation of the data, revising it for intellectual content and final approval of the version to be published. All authors have read and agreed to the published version of the manuscript.
Funding
This research project was supported by a grant from the South African Medical Research Council (SAMRC), with funds from National Treasury under its Economic Competitiveness and Support Package (MRC-RFA-UFSP-01-2013/VMH Study), South African National Research Foundation (SANRF) (Grant no. 115450). Any opinions, findings, conclusions, or recommendations expressed in this article are those of the author(s), and the SAMRC and/or SANRF do not accept any liability in this regard.
Acknowledgments
We thank the Bellville South community and their community Health Forum for supporting the study. T.E.M. takes full responsibility for the work as a whole, including the study design, access to data, and the decision to submit and publish the manuscript.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Deaton, A.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Luu, P.L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Hoarau-Vechot, J.; Fakhro, K.; Rafii, A.; Abi Khalil, C. Epigenetics and Cardiovascular Disease in Diabetes. Curr. Diab. Rep. 2015, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Pinney, S.E. DNA methylation and its role in the pathogenesis of diabetes. Pediatr. Diabetes 2017, 18, 167–177. [Google Scholar] [CrossRef]
- Muka, T.; Nano, J.; Voortman, T.; Braun, K.V.E.; Ligthart, S.; Stranges, S.; Bramer, W.M.; Troup, J.; Chowdhury, R.; Dehghan, A.; et al. The role of global and regional DNA methylation and histone modifications in glycemic traits and type 2 diabetes: A systematic review. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 553–566. [Google Scholar] [CrossRef]
- Ronn, T.; Ling, C. DNA methylation as a diagnostic and therapeutic target in the battle against Type 2 diabetes. Epigenomics 2015, 7, 451–460. [Google Scholar] [CrossRef]
- Li, Y.; Xu, K.; Xu, K.; Chen, S.; Cao, Y.; Zhan, H. Roles of Identified Long Noncoding RNA in Diabetic Nephropathy. J. Diabetes Res. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, X.; Wu, Z.; Hu, D.; Jia, J.; Guo, J.; Tang, T.; Yao, J.; Liu, H.; Tang, H. Long noncoding RNA LINC00467 promotes glioma progression through inhibiting p53 expression via binding to DNMT1. J. Cancer 2020, 11, 2935–2944. [Google Scholar] [CrossRef]
- Huang, Y.; Li, J.; Chen, S.; Zhao, S.; Huang, J.; Zhou, J.; Xu, Y. Identification of Potential Therapeutic Targets and Pathways of Liraglutide Against Type 2 Diabetes Mellitus (T2DM) Based on Long Non-Coding RNA (lncRNA) Sequencing. Med. Sci. Monit. 2020, 26, e922210. [Google Scholar] [CrossRef]
- Zhang, W.; Zheng, J.; Hu, X.; Chen, L. Dysregulated expression of long noncoding RNAs serves as diagnostic biomarkers of type 2 diabetes mellitus. Endocrine 2019, 65, 494–503. [Google Scholar] [CrossRef]
- Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. Linking a role of lncRNAs (long non-coding RNAs) with insulin resistance, accelerated senescence, and inflammation in patients with type 2 diabetes. Hum. Genomics 2018, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Leti, F.; DiStefano, J.K. Long noncoding RNAs as diagnostic and therapeutic targets in type 2 diabetes and related complications. Genes 2017, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Priya, G.; Kalra, S. Metformin in the management of diabetes during pregnancy and lactation. Drugs Context 2018, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichael, G.G.; Taylor, H.S.; et al. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene 2017, 36, 2345–2354. [Google Scholar] [CrossRef]
- Ishikawa, K.; Tsunekawa, S.; Ikeniwa, M.; Izumoto, T.; Iida, A.; Ogata, H.; Uenishi, E.; Seino, Y.; Ozaki, N.; Sugimura, Y.; et al. Long-term pancreatic beta cell exposure to high levels of glucose but not palmitate induces DNA methylation within the insulin gene promoter and represses transcriptional activity. PLoS ONE 2015, 10, e0115350. [Google Scholar] [CrossRef]
- García-Calzón, S.; Perfilyev, A.; Männistö, V.; de Mello, V.D.; Nilsson, E.; Pihlajamäki, J.; Ling, C. Diabetes medication associates with DNA methylation of metformin transporter genes in the human liver. Clin. Epigenetics 2017, 9, 1–9. [Google Scholar] [CrossRef]
- Elbere, I.; Silamikelis, I.; Ustinova, M.; Kalnina, I.; Zaharenko, L.; Peculis, R.; Konrade, I.; Ciuculete, D.M.; Zhukovsky, C.; Gudra, D.; et al. Significantly altered peripheral blood cell DNA methylation profile as a result of immediate effect of metformin use in healthy individuals. Clin. Epigenetics 2018, 10, 156. [Google Scholar] [CrossRef]
- Banerjee, P.; Surendran, H.; Chowdhury, D.R.; Prabhakar, K.; Pal, R. Metformin mediated reversal of epithelial to mesenchymal transition is triggered by epigenetic changes in E-cadherin promoter. J. Mol. Med. 2016, 94, 1397–1409. [Google Scholar] [CrossRef]
- Yu, X.; Mao, W.; Zhai, Y.; Tong, C.; Liu, M.; Ma, L.; Yu, X.; Li, S. Anti-tumor activity of metformin: From metabolic and epigenetic perspectives. Oncotarget 2017, 8, 5619–5628. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.F.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Sun, K.; Meng, Z.; Chen, L. The SLC transporter in nutrient and metabolic sensing, regulation, and drug development. J. Mol. Cell Biol. 2018, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Haitina, T.; Lindblom, J.; Renström, T.; Fredriksson, R. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics 2006, 88, 779–790. [Google Scholar] [CrossRef]
- Rodríguez-Mulero, S.; Errasti-Murugarren, E.; Ballarín, J.; Felipe, A.; Doucet, A.; Casado, F.J.; Pastor-Anglada, M. Expression of concentrative nucleoside transporters SLC28 (CNT1, CNT2, and CNT3) along the rat nephron: Effect of diabetes. Kidney Int. 2005, 68, 665–672. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Aspects Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Palmieri, F.; Monné, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta—Mol. Cell Res. 2016, 1863, 2362–2378. [Google Scholar] [CrossRef]
- Guo, K.; Elzinga, S.; Eid, S.; Figueroa-Romero, C.; Hinder, L.M.; Pacut, C.; Feldman, E.L.; Hur, J. Genome-wide DNA methylation profiling of human diabetic peripheral neuropathy in subjects with type 2 diabetes mellitus. Epigenetics 2019, 14, 766–779. [Google Scholar] [CrossRef]
- Fernyhough, P. Mitochondrial Dysfunction in Diabetic Neuropathy: A Series of Unfortunate Metabolic Events. Curr. Diab. Rep. 2015, 15, 24–27. [Google Scholar] [CrossRef]
- Fujimaki, S.; Kuwabara, T. Diabetes-induced dysfunction of mitochondria and stem cells in skeletal muscle and the nervous system. Int. J. Mol. Sci. 2017, 18, 2147. [Google Scholar] [CrossRef]
- Pop-Busui, R.; Lu, J.; Brooks, M.M.; Albert, S.; Althouse, A.D.; Escobedo, J.; Green, J.; Palumbo, P.; Perkins, B.A.; Whitehouse, F.; et al. Impact of glycemic control strategies ontheprogressionofdiabeticperipheral neuropathy in the bypass angioplasty revascularization investigation 2 diabetes (BARI 2D) Cohort. Diabetes Care 2013, 36, 3208–3215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Lin, J.D. Long Noncoding RNAs: A New Regulatory Code in Metabolic Control. Trends Biochem. Sci. 2015, 40, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.H.; Kaur, S.; Brorsson, C.A.; Pociot, F. Effects of GWAS-associated genetic variants on lncRNAs within IBD and T1D candidate loci. PLoS ONE 2014, 9, e105723. [Google Scholar] [CrossRef] [PubMed]
- Pengyu, Z.; Yan, Y.; Xiying, F.; Maoguang, Y.; Mo, L.; Yan, C.; Hong, S.; Lijuan, W.; Xiujuan, Z.; Hanqing, C. The Differential Expression of Long Noncoding RNAs in Type 2 Diabetes Mellitus and Latent Autoimmune Diabetes in Adults. Int. J. Endocrinol. 2020, 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Kim, Y.; Caramori, M.L.; Moore, J.H.; Rich, S.S.; Mychaleckyj, J.C.; Walker, P.C.; Mauer, M. Diabetic nephropathy is associated with gene expression levels of oxidative phosphorylation and related pathways. Diabetes 2006, 55, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Farré, P.; Jones, M.J.; Meaney, M.J.; Emberly, E.; Turecki, G.; Kobor, M.S. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin 2015, 8, 1–17. [Google Scholar] [CrossRef]
- Crujeiras, A.B.; Diaz-Lagares, A.; Sandoval, J.; Milagro, F.I.; Navas-Carretero, S.; Carreira, M.C.; Gomez, A.; Hervas, D.; Monteiro, M.P.; Casanueva, F.F.; et al. DNA methylation map in circulating leukocytes mirrors subcutaneous adipose tissue methylation pattern: A genome-wide analysis from non-obese and obese patients. Sci. Rep. 2017, 7, 41903. [Google Scholar] [CrossRef]
- Porta, M.; Curletto, G.; Cipullo, D.; De la Longrais, R.R.; Trento, M.; Passera, P.; Taulaigo, A.V.; Di Miceli, S.; Cenci, A.; Dalmasso, P.; et al. Estimating the Delay Between Onset and Diagnosis of Type 2 Diabetes From the Time Course of Retinopathy Prevalence. Diabetes Care 2014, 37, 1668–1674. [Google Scholar] [CrossRef]
- Down, T.A.; Rakyan, V.K.; Turner, D.J.; Flicek, P.; Li, H.; Kulesha, E.; Gräf, S.; Johnson, N.; Herrero, J.; Tomazou, E.M.; et al. A Bayesian deconvolution strategy for immunoprecipitation- based DNA methylome analysis. Nat. Biotechnol. 2008, 26, 779–785. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).