
Deciphering Plant Chromatin Regulation via CRISPR/dCas9-Based Epigenome Engineering


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Advances in CRISPR-dCas9 Epigenome Editing in Plants
2.1. Transcriptional Engineering in Plants
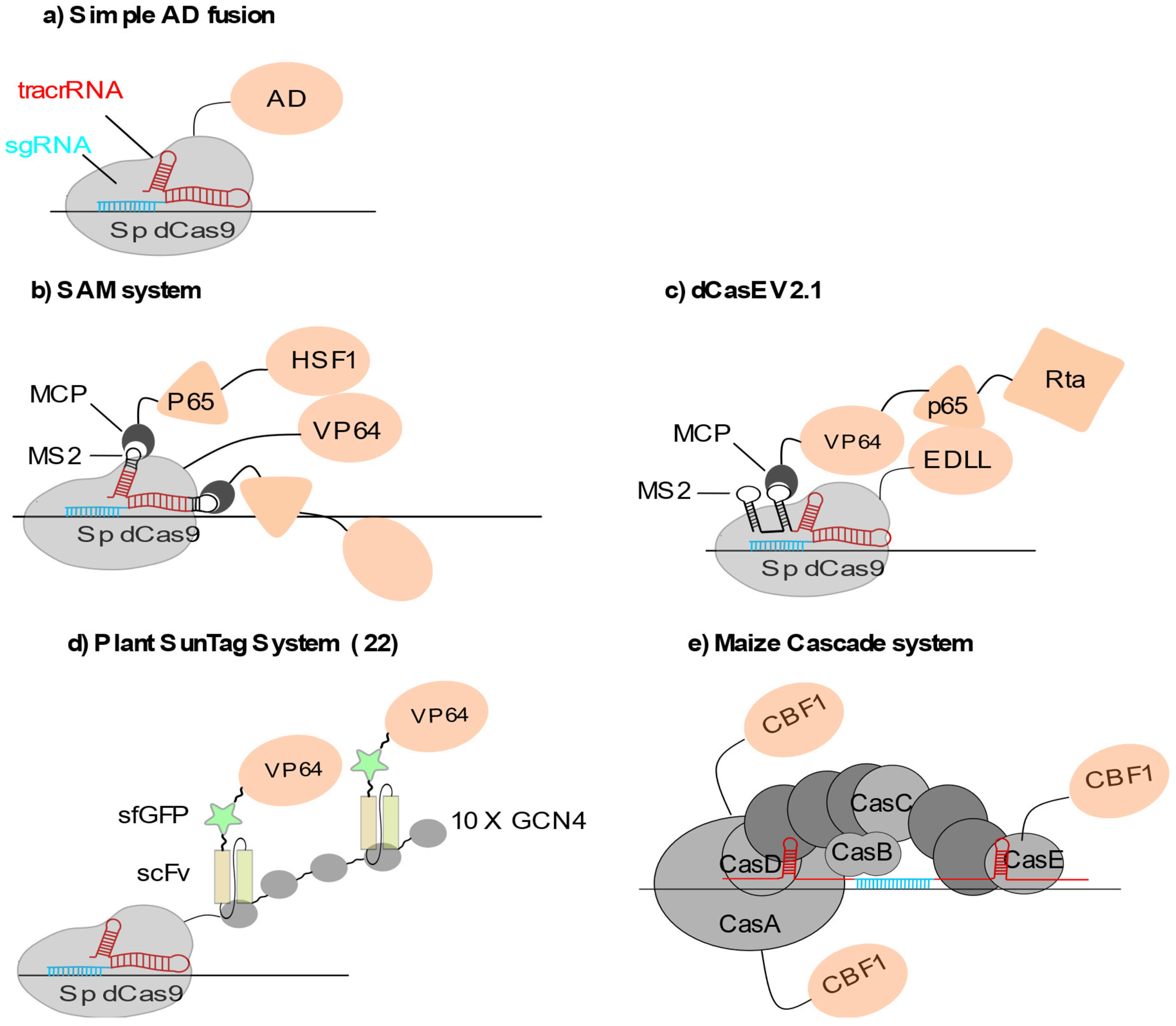
2.1.1. Locus-Specific Transcriptional Activation (CRISPRa)
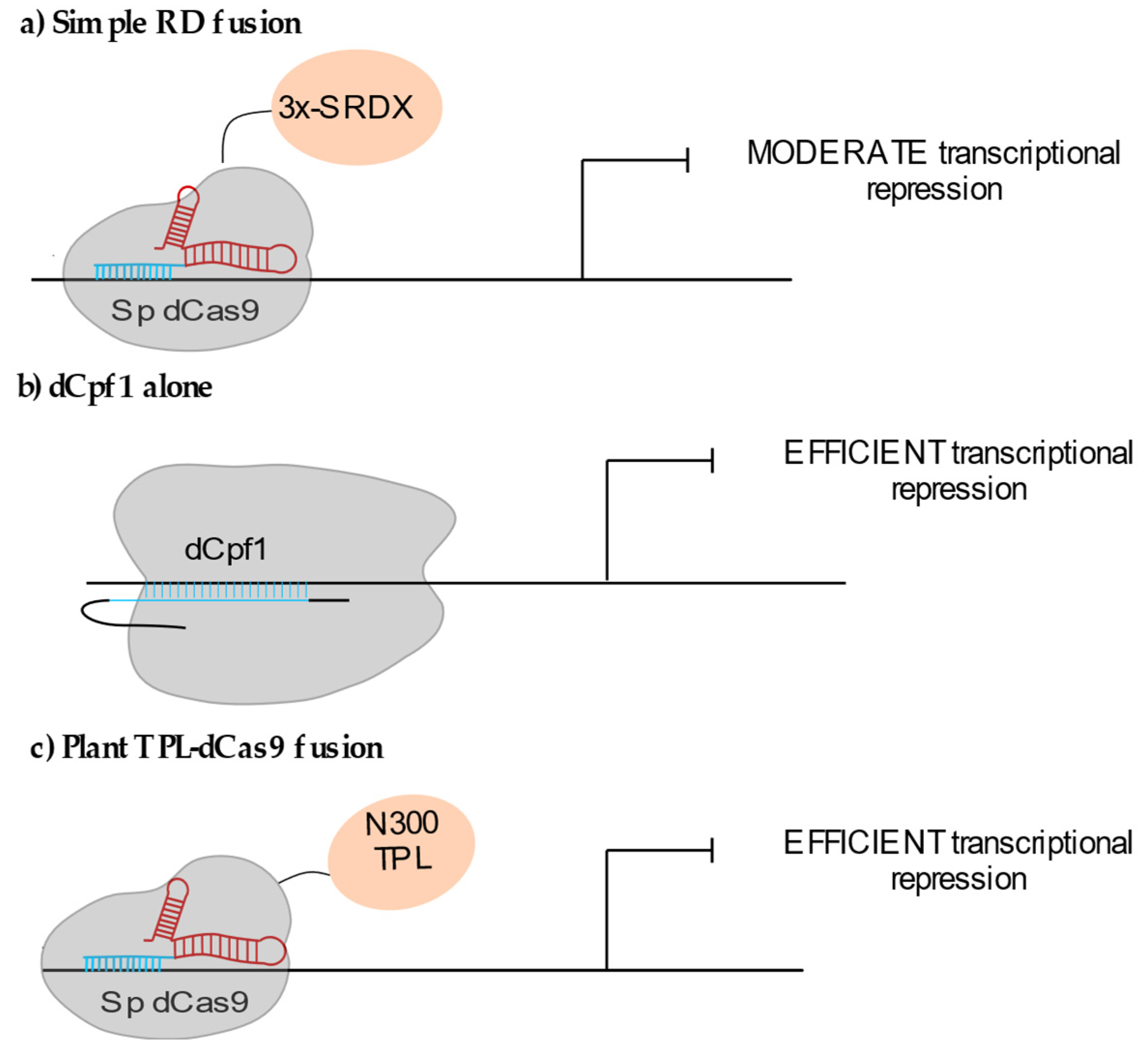
2.1.2. Locus-Specific Transcriptional Repression (CRISPRi)
2.2. Epigenome Editing to Manipulate Plant Chromatin Homeostasis
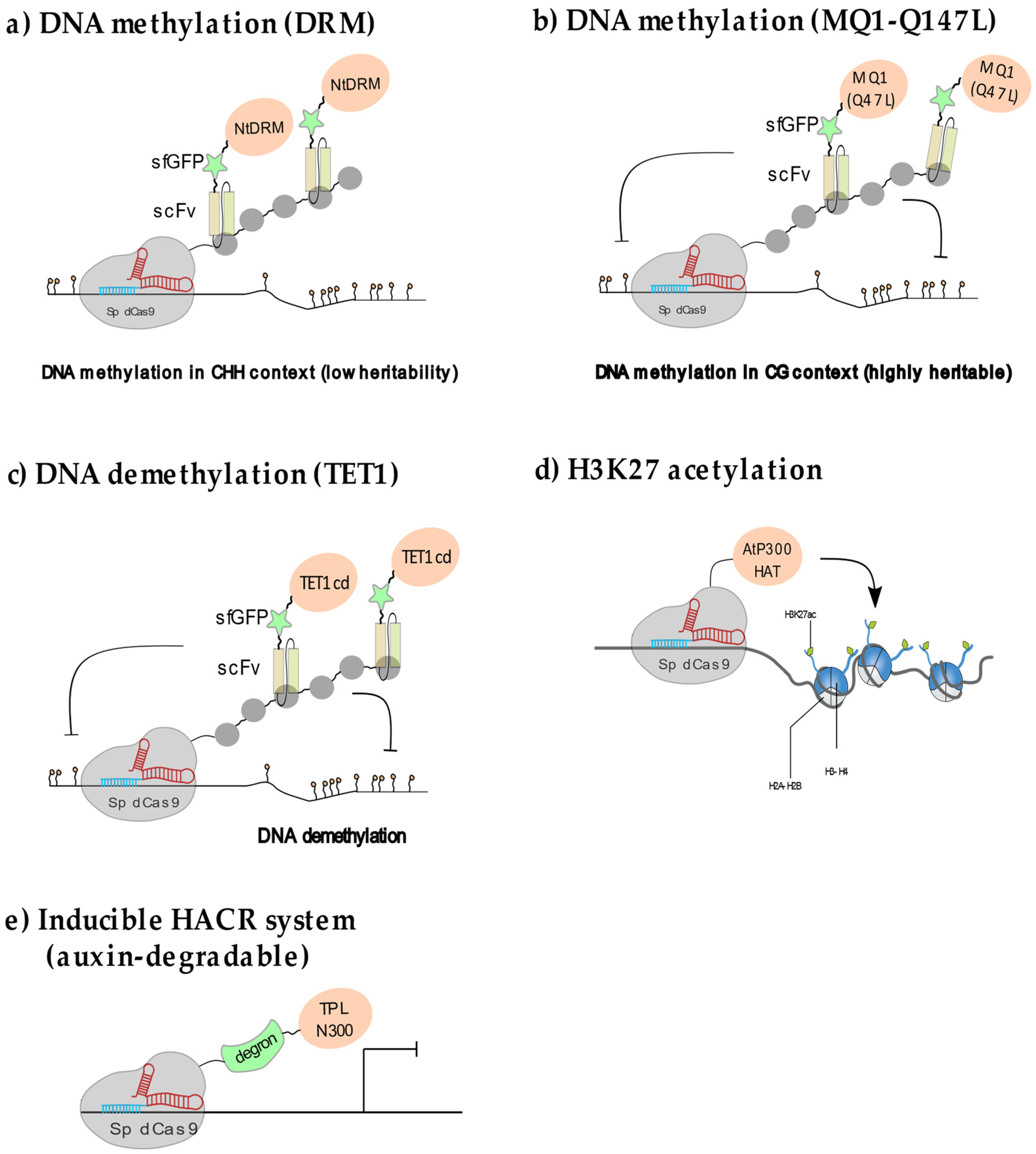
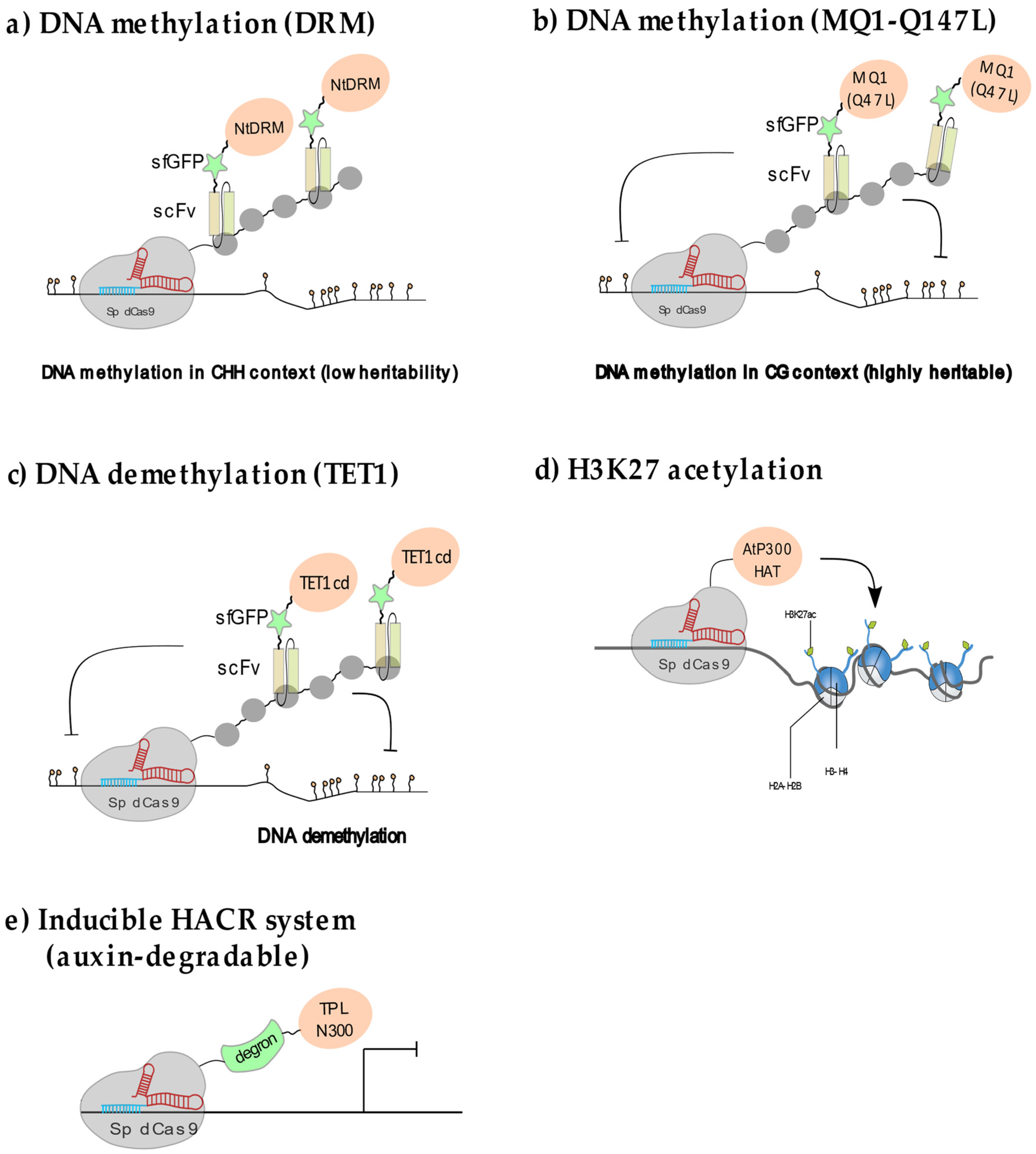
2.2.1. Manipulating Plant DNA Methylation
2.2.2. Locus-Specific Demethylation in Plants
2.2.3. Manipulating Plant Histone Acetylation and Methylation
3. Epigenome Editing to Interrogate the Effect of Chromatin Regulator Interactions on Transcription
3.1. Positive Crosstalks between Chromatin Regulator Pathways
3.2. Complex Interactions between Chromatin Regulatory Functions
3.2.1. Modulation of H3K27 Methylation Reveal Cooperative as Well as Antagonistic Interactions
3.2.2. Antagonism between DNA Methylation and Transcription Factors
3.3. Transcription Factors and Histone Acetyltransferases Have Distinct Effects on the Timing of Transcription
4. Exploiting Current Limitations in Using CRISPR/dCas9 Approaches and Going beyond
4.1. Locus Accessibility and Chromatin Status Impacts on CRISPR/dCas9 Output
4.2. Cell-Type Specific Responses to Targeted Chromatin Modifiers
4.3. Combining Epigenome Editing with Cell-Specific Approaches
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Talbert, P.B.; Meers, M.P.; Henikoff, S. Old Cogs, New Tricks: The Evolution of Gene Expression in a Chromatin Context. Nat. Rev. Genet. 2019, 20, 283–297. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. The Yin and Yang of Histone Marks in Transcription. Annu. Rev. Genom. Hum. Genet. 2021, 22, 1. [Google Scholar] [CrossRef] [PubMed]
- Rath, D.; Amlinger, L.; Rath, A.; Lundgren, M. The CRISPR-Cas Immune System: Biology, Mechanisms and Applications. Biochimie 2015, 117, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Young, J.K.; Gasior, S.L.; Jones, S.; Wang, L.; Navarro, P.; Vickroy, B.; Barrangou, R. The Repurposing of Type I-E CRISPR-Cascade for Gene Activation in Plants. Commun. Biol. 2019, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Fal, K.; Tomkova, D.; Vachon, G.; Chabouté, M.-E.; Berr, A.; Carles, C.C. Chromatin Manipulation and Editing: Challenges, New Technologies and Their Use in Plants. Int. J. Mol. Sci. 2021, 22, 512. [Google Scholar] [CrossRef]
- Zhang, Y.; Malzahn, A.A.; Sretenovic, S.; Qi, Y. The Emerging and Uncultivated Potential of CRISPR Technology in Plant Science. Nat. Plants 2019, 5, 778–794. [Google Scholar] [CrossRef] [PubMed]
- Selma, S.; Bernabé-Orts, J.M.; Vazquez-Vilar, M.; Diego-Martin, B.; Ajenjo, M.; Garcia-Carpintero, V.; Granell, A.; Orzaez, D. Strong Gene Activation in Plants with Genome-wide Specificity Using a New Orthogonal CRISPR /Cas9-based Programmable Transcriptional Activator. Plant Biotechnol. J. 2019, 17, 1703–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.; Schuetz, T.J.; Sullivan, E.K.; Kingston, R.E. A Heat Shock-Responsive Domain of Human HSF1 That Regulates Transcription Activation Domain Function. Mol. Cell. Biol. 1995, 15, 3354–3362. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.B.; Belachew, A.; Ma, S.F.; Young, M.; Ade, J.; Shen, Y.; Marion, C.M.; Holtan, H.E.; Bailey, A.; Stone, J.K.; et al. The EDLL Motif: A Potent Plant Transcriptional Activation Domain from AP2/ERF Transcription Factors. Plant J. Cell Mol. Biol. 2012, 70, 855–865. [Google Scholar] [CrossRef]
- Lowder, L.G.; Zhang, D.; Baltes, N.J.; Paul, J.W.; Tang, X.; Zheng, X.; Voytas, D.F.; Hsieh, T.-F.; Zhang, Y.; Qi, Y. A CRISPR/Cas9 Toolbox for Multiplexed Plant Genome Editing and Transcriptional Regulation. Plant Physiol. 2015, 169, 971–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piatek, A.; Ali, Z.; Baazim, H.; Li, L.; Abulfaraj, A.; Al-Shareef, S.; Aouida, M.; Mahfouz, M.M. RNA-Guided Transcriptional Regulation in Planta via Synthetic DCas9-Based Transcription Factors. Plant Biotechnol. J. 2015, 13, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Choy, B.; Green, M.R. Eukaryotic Activators Function during Multiple Steps of Preinitiation Complex Assembly. Nature 1993, 366, 531–536. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome Editing by a CRISPR-Cas9-Based Acetyltransferase Activates Genes from Promoters and Enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-Scale Transcriptional Activation by an Engineered CRISPR-Cas9 Complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, H.E.; Liljas, L.; Uhlenbeck, O.C. RNA Recognition by the MS2 Phage Coat Protein. Semin. Virol. 1997, 8, 176–185. [Google Scholar] [CrossRef]
- Lim, F.; Peabody, D.S. RNA Recognition Site of PP7 Coat Protein. Nucleic Acids Res. 2002, 30, 4138–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, A.; Tuttle, M.; Pruitt, B.W.; Ewen-Campen, B.; Chari, R.; Ter-Ovanesyan, D.; Haque, S.J.; Cecchi, R.J.; Kowal, E.J.K.; Buchthal, J.; et al. Comparison of Cas9 Activators in Multiple Species. Nat. Methods 2016, 13, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, D.; Xiong, X.; Yan, B.; Xie, W.; Sheen, J.; Li, J.-F. A Potent Cas9-Derived Gene Activator for Plant and Mammalian Cells. Nat. Plants 2017, 3, 930–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowder, L.G.; Zhou, J.; Zhang, Y.; Malzahn, A.; Zhong, Z.; Hsieh, T.-F.; Voytas, D.F.; Zhang, Y.; Qi, Y. Robust Transcriptional Activation in Plants Using Multiplexed CRISPR-Act2.0 and MTALE-Act Systems. Mol. Plant 2018, 11, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-Specific Manipulation of Arabidopsis Loci Using CRISPR-Cas9 SunTag Systems. Nat. Commun. 2019, 10, 729. [Google Scholar] [CrossRef]
- Brouns, S.J.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.H.; Snijders, A.P.L.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [Green Version]
- Jore, M.M.; Lundgren, M.; van Duijn, E.; Bultema, J.B.; Westra, E.R.; Waghmare, S.P.; Wiedenheft, B.; Pul, Ü.; Wurm, R.; Wagner, R.; et al. Structural Basis for CRISPR RNA-Guided DNA Recognition by Cascade. Nat. Struct. Mol. Biol. 2011, 18, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Vilar, M.; Bernabé-Orts, J.M.; Fernandez-del-Carmen, A.; Ziarsolo, P.; Blanca, J.; Granell, A.; Orzaez, D. A Modular Toolbox for GRNA–Cas9 Genome Engineering in Plants Based on the GoldenBraid Standard. Plant Methods 2016, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Hiratsu, K.; Matsui, K.; Koyama, T.; Ohme-Takagi, M. Dominant Repression of Target Genes by Chimeric Repressors That Include the EAR Motif, a Repression Domain, in Arabidopsis. Plant J. Cell Mol. Biol. 2003, 34, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Mahfouz, M.M.; Li, L.; Piatek, M.; Fang, X.; Mansour, H.; Bangarusamy, D.K.; Zhu, J.-K. Targeted Transcriptional Repression Using a Chimeric TALE-SRDX Repressor Protein. Plant Mol. Biol. 2012, 78, 311–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Lowder, L.G.; Zhang, T.; Malzahn, A.A.; Zheng, X.; Voytas, D.F.; Zhong, Z.; Chen, Y.; Ren, Q.; Li, Q.; et al. A CRISPR-Cpf1 System for Efficient Genome Editing and Transcriptional Repression in Plants. Nat. Plants 2017, 3, 17018. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Ren, Q.; Pan, C.; He, Y.; Zhang, Y.; Liu, S.; Zhong, Z.; Wang, J.; Malzahn, A.A.; Wu, J.; et al. CRISPR–Cas12b Enables Efficient Plant Genome Engineering. Nat. Plants 2020, 6, 202–208. [Google Scholar] [CrossRef]
- Swarts, D.C.; van der Oost, J.; Jinek, M. Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Mol. Cell 2017, 66, 221–233.e4. [Google Scholar] [CrossRef] [Green Version]
- Khakhar, A.; Leydon, A.R.; Lemmex, A.C.; Klavins, E.; Nemhauser, J.L. Synthetic Hormone-Responsive Transcription Factors Can Monitor and Re-Program Plant Development. eLife 2018, 7, e34702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leydon, A.R.; Wang, W.; Gala, H.P.; Gilmour, S.; Juarez-Solis, S.; Zahler, M.L.; Zemke, J.E.; Zheng, N.; Nemhauser, J.L. Repression by the Arabidopsis TOPLESS Corepressor Requires Association with the Core Mediator Complex. eLife 2021, 10, e66739. [Google Scholar] [CrossRef] [PubMed]
- Long, J.A.; Ohno, C.; Smith, Z.R.; Meyerowitz, E.M. TOPLESS Regulates Apical Embryonic Fate in Arabidopsis. Science 2006, 312, 1520–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and Function of DNA Methylation in Plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J. DNA Methylation in Plants: Mechanisms and Tools for Targeted Manipulation. New Phytol. 2020, 227, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, E.; Grimmer, M.R.; Stolzenburg, S.; Bogdanovic, O.; de Mendoza, A.; Farnham, P.J.; Blancafort, P.; Lister, R. Frequent Lack of Repressive Capacity of Promoter DNA Methylation Identified through Genome-Wide Epigenomic Manipulation. bioRxiv 2017, 170506. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Zhang, X.; Su, J.; Jeong, M.; Gundry, M.C.; Huang, Y.-H.; Zhou, Y.; Li, W.; Goodell, M.A. Targeted DNA Methylation in Vivo Using an Engineered DCas9-MQ1 Fusion Protein. Nat. Commun. 2017, 8, 16026. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Gallego-Bartolomé, J.; Zhou, Y.; Zhong, Z.; Wang, M.; Wongpalee, S.P.; Gardiner, J.; Feng, S.; Kuo, P.H.; Jacobsen, S.E. Ectopic Targeting of CG DNA Methylation in Arabidopsis with the Bacterial SssI Methyltransferase. Nat. Commun. 2021, 12, 3130. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, B.; Picard, C.L.; Vong, B.; Feng, S.; Jacobsen, S.E. CRISPR-Based Targeting of DNA Methylation in Arabidopsis Thaliana by a Bacterial CG-Specific DNA Methyltransferase. Proc. Natl. Acad. Sci. USA 2021, 118, e2125016118. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y. TET-Mediated Active DNA Demethylation: Mechanism, Function and Beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.-C.; Segal, D.J.; Jacobsen, S.E. Targeted DNA Demethylation of the Arabidopsis Genome Using the Human TET1 Catalytic Domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125–E2134. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.V.; Bassal, M.A.; Lin, Q.X.X.; Wu, C.-S.; Kwon, J.; Zhou, Q.; Tan, H.K.; Ebralidze, A.K.; Chai, L.; Benoukraf, T.; et al. Targeted Intragenic Demethylation Initiates Chromatin Rewiring for Gene Activation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, P.; Chen, M.; Liu, Y.; Qi, L.S.; Ding, S. CRISPR-Based Chromatin Remodeling of the Endogenous Oct4 or Sox2 Locus Enables Reprogramming to Pluripotency. Cell Stem Cell 2018, 22, 252–261.e4. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Neumann, M.; Duro, D.I.; Schmid, M. CRISPR-Based Tools for Targeted Transcriptional and Epigenetic Regulation in Plants. PLoS ONE 2019, 14, e0222778. [Google Scholar] [CrossRef]
- Roca Paixão, J.F.; Gillet, F.-X.; Ribeiro, T.P.; Bournaud, C.; Lourenço-Tessutti, I.T.; Noriega, D.D.; de Melo, B.P.; de Almeida-Engler, J.; Grossi-de-Sa, M.F. Improved Drought Stress Tolerance in Arabidopsis by CRISPR/DCas9 Fusion with a Histone AcetylTransferase. Sci. Rep. 2019, 9, 8080. [Google Scholar] [CrossRef] [Green Version]
- de Melo, B.P.; Lourenço-Tessutti, I.T.; Paixão, J.F.R.; Noriega, D.D.; Silva, M.C.M.; de Almeida-Engler, J.; Fontes, E.P.B.; Grossi-de-Sa, M.F. Transcriptional Modulation of AREB-1 by CRISPRa Improves Plant Physiological Performance under Severe Water Deficit. Sci. Rep. 2020, 10, 16231. [Google Scholar] [CrossRef]
- Hu, J.; Lei, Y.; Wong, W.-K.; Liu, S.; Lee, K.-C.; He, X.; You, W.; Zhou, R.; Guo, J.-T.; Chen, X.; et al. Direct Activation of Human and Mouse Oct4 Genes Using Engineered TALE and Cas9 Transcription Factors. Nucleic Acids Res. 2014, 42, 4375–4390. [Google Scholar] [CrossRef] [PubMed]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, N.C.; Chavez, A.; Lance-Byrne, A.; Chan, Y.; Menn, D.; Milanova, D.; Kuo, C.-C.; Guo, X.; Sharma, S.; Tung, A.; et al. An Enhanced CRISPR Repressor for Targeted Mammalian Gene Regulation. Nat. Methods 2018, 15, 611–616. [Google Scholar] [CrossRef]
- Carlson-Stevermer, J.; Kelso, R.; Kadina, A.; Joshi, S.; Rossi, N.; Walker, J.; Stoner, R.; Maures, T. CRISPRoff Enables Spatio-Temporal Control of CRISPR Editing. Nat. Commun. 2020, 11, 5041. [Google Scholar] [CrossRef]
- Vergara, Z.; Gutierrez, C. Emerging Roles of Chromatin in the Maintenance of Genome Organization and Function in Plants. Genome Biol. 2017, 18, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano-Rodriguez, D.; Gjaltema, R.A.F.; Jilderda, L.J.; Jellema, P.; Dokter-Fokkens, J.; Ruiters, M.H.J.; Rots, M.G. Writing of H3K4Me3 Overcomes Epigenetic Silencing in a Sustained but Context-Dependent Manner. Nat. Commun. 2016, 7, 12284. [Google Scholar] [CrossRef]
- Zhang, K.; Sridhar, V.V.; Zhu, J.; Kapoor, A.; Zhu, J.-K. Distinctive Core Histone Post-Translational Modification Patterns in Arabidopsis Thaliana. PLoS ONE 2007, 2, e1210. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, A.-S.; Bourbousse, C.; Concia, L.; Rougée, M.; Deton-Cabanillas, A.-F.; Zabulon, G.; Layat, E.; Latrasse, D.; Kim, S.K.; Chaumont, N.; et al. Arabidopsis S2Lb Links AtCOMPASS-like and SDG2 Activity in H3K4me3 Independently from Histone H2B Monoubiquitination. Genome Biol. 2019, 20, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugues, A.; Jacobs, C.S.; Roudier, F. Mitotic Inheritance of PRC2-Mediated Silencing: Mechanistic Insights and Developmental Perspectives. Front. Plant Sci. 2020, 11, 262. [Google Scholar] [CrossRef] [Green Version]
- O’Geen, H.; Ren, C.; Nicolet, C.M.; Perez, A.A.; Halmai, J.; Le, V.M.; Mackay, J.P.; Farnham, P.J.; Segal, D.J. DCas9-Based Epigenome Editing Suggests Acquisition of Histone Methylation Is Not Sufficient for Target Gene Repression. Nucleic Acids Res. 2017, 45, 9901–9916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, H.S.; Takeda, H.; Nakamura, R. Targeted in Vivo Epigenome Editing of H3K27me3. Epigenet. Chromatin 2019, 12, 17. [Google Scholar] [CrossRef]
- O’Geen, H.; Bates, S.L.; Carter, S.S.; Nisson, K.A.; Halmai, J.; Fink, K.D.; Rhie, S.K.; Farnham, P.J.; Segal, D.J. Ezh2-DCas9 and KRAB-DCas9 Enable Engineering of Epigenetic Memory in a Context-Dependent Manner. Epigenet. Chromatin 2019, 12, 26. [Google Scholar] [CrossRef]
- Bartke, T.; Vermeulen, M.; Xhemalce, B.; Robson, S.C.; Mann, M.; Kouzarides, T. Nucleosome-Interacting Proteins Regulated by DNA and Histone Methylation. Cell 2010, 143, 470–484. [Google Scholar] [CrossRef] [Green Version]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-Scale DNA Methylation Maps of Pluripotent and Differentiated Cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA Methylomes at Base Resolution Show Widespread Epigenomic Differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Clarenz, O.; Cokus, S.; Bernatavichute, Y.V.; Pellegrini, M.; Goodrich, J.; Jacobsen, S.E. Whole-Genome Analysis of Histone H3 Lysine 27 Trimethylation in Arabidopsis. PLoS Biol. 2007, 5, e129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinhofer, I.; Hehenberger, E.; Roszak, P.; Hennig, L.; Köhler, C. H3K27me3 Profiling of the Endosperm Implies Exclusion of Polycomb Group Protein Targeting by DNA Methylation. PLoS Genet. 2010, 6, e1001152. [Google Scholar] [CrossRef]
- Moody, J.D.; Levy, S.; Mathieu, J.; Xing, Y.; Kim, W.; Dong, C.; Tempel, W.; Robitaille, A.M.; Dang, L.T.; Ferreccio, A.; et al. First Critical Repressive H3K27me3 Marks in Embryonic Stem Cells Identified Using Designed Protein Inhibitor. Proc. Natl. Acad. Sci. USA 2017, 114, 10125–10130. [Google Scholar] [CrossRef] [Green Version]
- Levy, S.; Somasundaram, L.; Raj, I.X.; Ic-Mex, D.; Schmidt, S.; Alghadeer, A.; Honkanen, H.; Hawkins, R.D.; Mathieu, J.; Wang, Y.; et al. Computer Designed PRC2 Inhibitor, EBdCas9, Reveals Functional TATA Boxes in Distal Promoter Regions. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, Y.; Loh, Y.P.; Tng, J.Q.; Lim, M.C.; Cao, Z.; Raju, A.; Aiden, E.L.; Li, S.; Manikandan, L.; et al. H3K27me3-Rich Genomic Regions Can Function as Silencers to Repress Gene Expression via Chromatin Interactions. Nat. Commun. 2021, 12, 719. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Ortega, J.A.; Quinlan, K.A.; Santos, D.P.; Gu, H.; Martin, E.J.; Galonska, C.; Pop, R.; Maidl, S.; Di Pardo, A.; et al. Dissecting the Functional Consequences of De Novo DNA Methylation Dynamics in Human Motor Neuron Differentiation and Physiology. Cell Stem Cell 2018, 22, 559–574.e9. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, R.C.; Huang, S.-S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 2016, 165, 1280–1292. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.J.; Scheibe, M.; Wongpalee, S.P.; Liu, W.; Cornett, E.M.; Vaughan, R.M.; Li, X.; Chen, W.; Xue, Y.; Zhong, Z.; et al. A DNA Methylation Reader Complex That Enhances Gene Transcription. Science 2018, 362, 1182–1186. [Google Scholar] [CrossRef] [Green Version]
- Ichino, L.; Boone, B.A.; Strauskulage, L.; Harris, C.J.; Kaur, G.; Gladstone, M.A.; Tan, M.; Feng, S.; Jami-Alahmadi, Y.; Duttke, S.H.; et al. MBD5 and MBD6 Couple DNA Methylation to Gene Silencing through the J-Domain Protein SILENZIO. Science 2021, 372, 1434–1439. [Google Scholar] [CrossRef]
- Nicolas, D.; Zoller, B.; Suter, D.M.; Naef, F. Modulation of Transcriptional Burst Frequency by Histone Acetylation. Proc. Natl. Acad. Sci. USA 2018, 115, 7153–7158. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-F.; Lin, Y.T.; Gallegos, D.A.; Hazlett, M.F.; Gómez-Schiavon, M.; Yang, M.G.; Kalmeta, B.; Zhou, A.S.; Holtzman, L.; Gersbach, C.A.; et al. Enhancer Histone Acetylation Modulates Transcriptional Bursting Dynamics of Neuronal Activity-Inducible Genes. Cell Rep. 2019, 26, 1174–1188.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaac, R.S.; Jiang, F.; Doudna, J.A.; Lim, W.A.; Narlikar, G.J.; Almeida, R. Nucleosome Breathing and Remodeling Constrain CRISPR-Cas9 Function. eLife 2016, 5, e13450. [Google Scholar] [CrossRef] [PubMed]
- Yarrington, R.M.; Verma, S.; Schwartz, S.; Trautman, J.K.; Carroll, D. Nucleosomes Inhibit Target Cleavage by CRISPR-Cas9 in Vivo. Proc. Natl. Acad. Sci. USA 2018, 115, 9351–9358. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kuscu, C.; Quinlan, A.; Qi, Y.; Adli, M. Cas9-Chromatin Binding Information Enables More Accurate CRISPR off-Target Prediction. Nucleic Acids Res. 2015, 43, e118. [Google Scholar] [CrossRef] [PubMed]
- Uusi-Mäkelä, M.I.E.; Barker, H.R.; Bäuerlein, C.A.; Häkkinen, T.; Nykter, M.; Rämet, M. Chromatin Accessibility Is Associated with CRISPR-Cas9 Efficiency in the Zebrafish (Danio Rerio). PLoS ONE 2018, 13, e0196238. [Google Scholar] [CrossRef] [Green Version]
- Daer, R.; Barrett, C.M.; Haynes, K.A. Histone Modifications and Active Gene Expression Are Associated with Enhanced CRISPR Activity in De-Silenced Chromatin. bioRxiv 2018, 228601. [Google Scholar] [CrossRef] [Green Version]
- Baumann, V.; Wiesbeck, M.; Breunig, C.T.; Braun, J.M.; Köferle, A.; Ninkovic, J.; Götz, M.; Stricker, S.H. Targeted Removal of Epigenetic Barriers during Transcriptional Reprogramming. Nat. Commun. 2019, 10, 2119. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.; Tian, X.; Xu, J. Single-Cell Transcriptome Analysis in Plants: Advances and Challenges. Mol. Plant 2021, 14, 115–126. [Google Scholar] [CrossRef]
- Alamos, S.; Reimer, A.; Niyogi, K.K.; Garcia, H.G. Quantitative Imaging of RNA Polymerase II Activity in Plants Reveals the Single-Cell Basis of Tissue-Wide Transcriptional Dynamics. Nat. Plants 2021, 7, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.; Olsson, T.S.G.; Hartley, M.; Dean, C.; Rosa, S. A Method for Detecting Single MRNA Molecules in Arabidopsis Thaliana. Plant Methods 2016, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Decaestecker, W.; Buono, R.A.; Pfeiffer, M.L.; Vangheluwe, N.; Jourquin, J.; Karimi, M.; Isterdael, G.V.; Beeckman, T.; Nowack, M.K.; Jacobs, T.B. CRISPR-TSKO: A Technique for Efficient Mutagenesis in Specific Cell Types, Tissues, or Organs in Arabidopsis. Plant Cell 2019, 31, 2868–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ye, L.; Lyu, M.; Ursache, R.; Löytynoja, A.; Mähönen, A.P. An Inducible Genome Editing System for Plants. Nat. Plants 2020, 6, 766–772. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubois, A.; Roudier, F. Deciphering Plant Chromatin Regulation via CRISPR/dCas9-Based Epigenome Engineering. Epigenomes 2021, 5, 17. https://doi.org/10.3390/epigenomes5030017
Dubois A, Roudier F. Deciphering Plant Chromatin Regulation via CRISPR/dCas9-Based Epigenome Engineering. Epigenomes. 2021; 5(3):17. https://doi.org/10.3390/epigenomes5030017
Chicago/Turabian StyleDubois, Annick, and François Roudier. 2021. "Deciphering Plant Chromatin Regulation via CRISPR/dCas9-Based Epigenome Engineering" Epigenomes 5, no. 3: 17. https://doi.org/10.3390/epigenomes5030017
APA StyleDubois, A., & Roudier, F. (2021). Deciphering Plant Chromatin Regulation via CRISPR/dCas9-Based Epigenome Engineering. Epigenomes, 5(3), 17. https://doi.org/10.3390/epigenomes5030017