
Multiomics Data Analysis Identified CpG Sites That Mediate the Impact of Smoking on Cardiometabolic Traits

Abstract
:1. Introduction
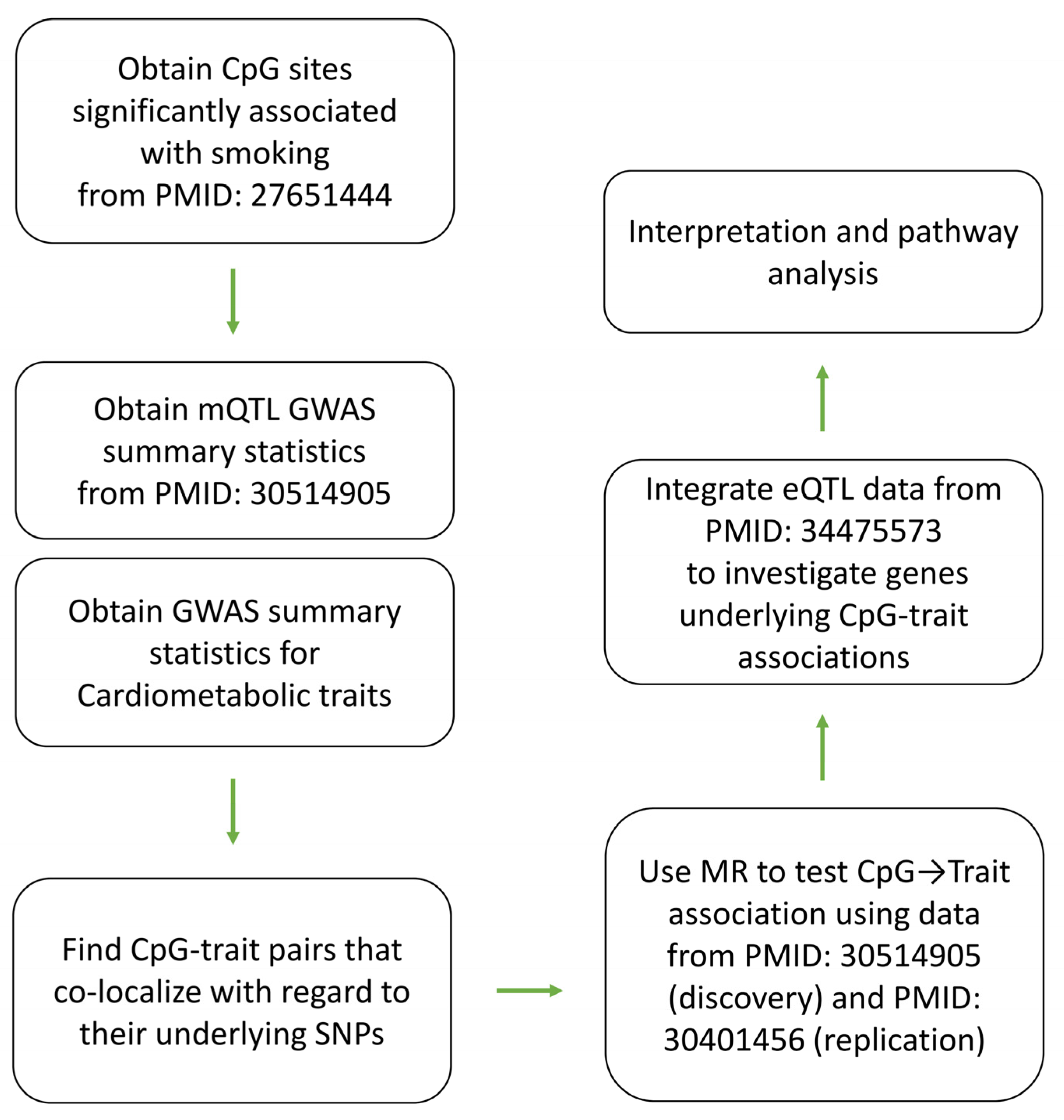
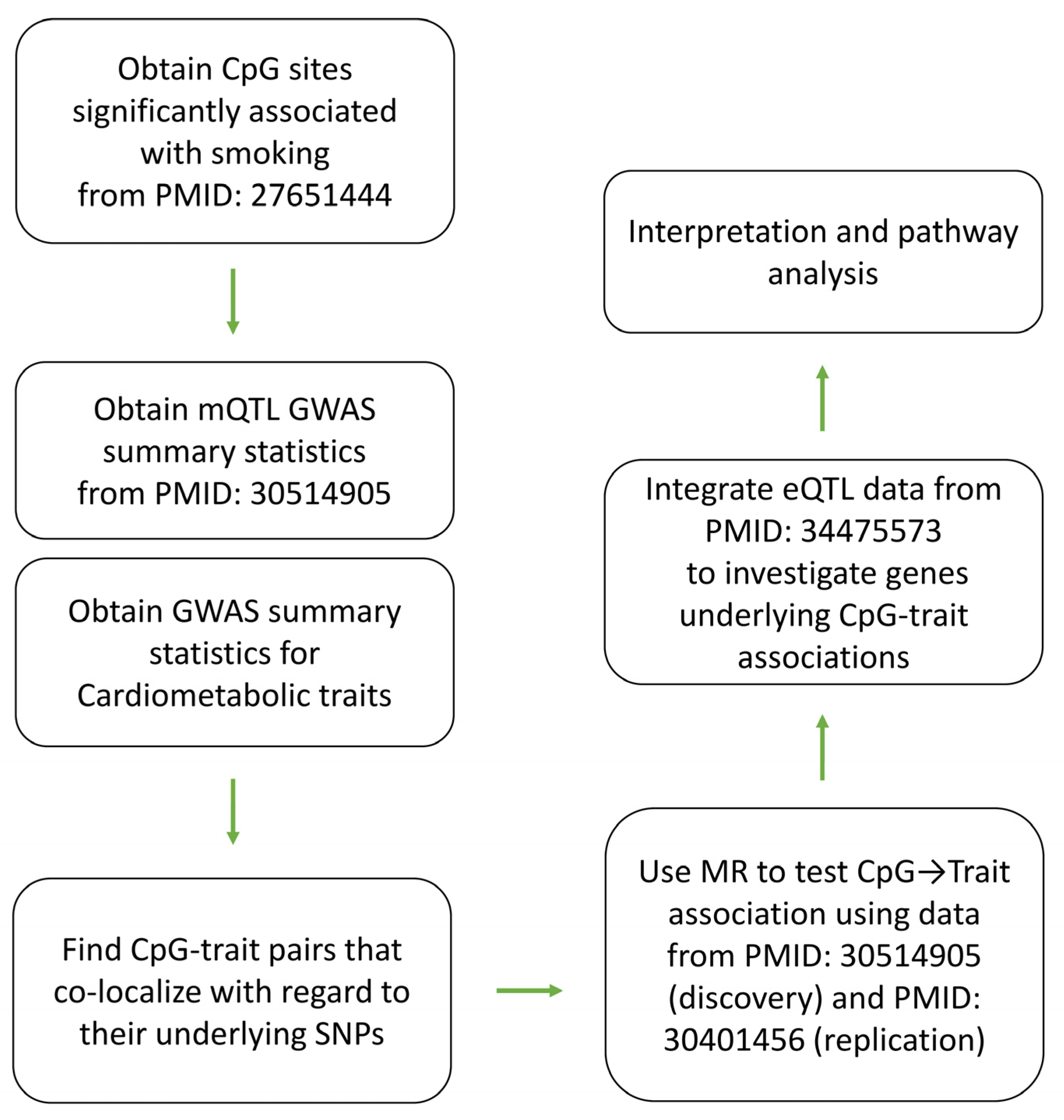
2. Results
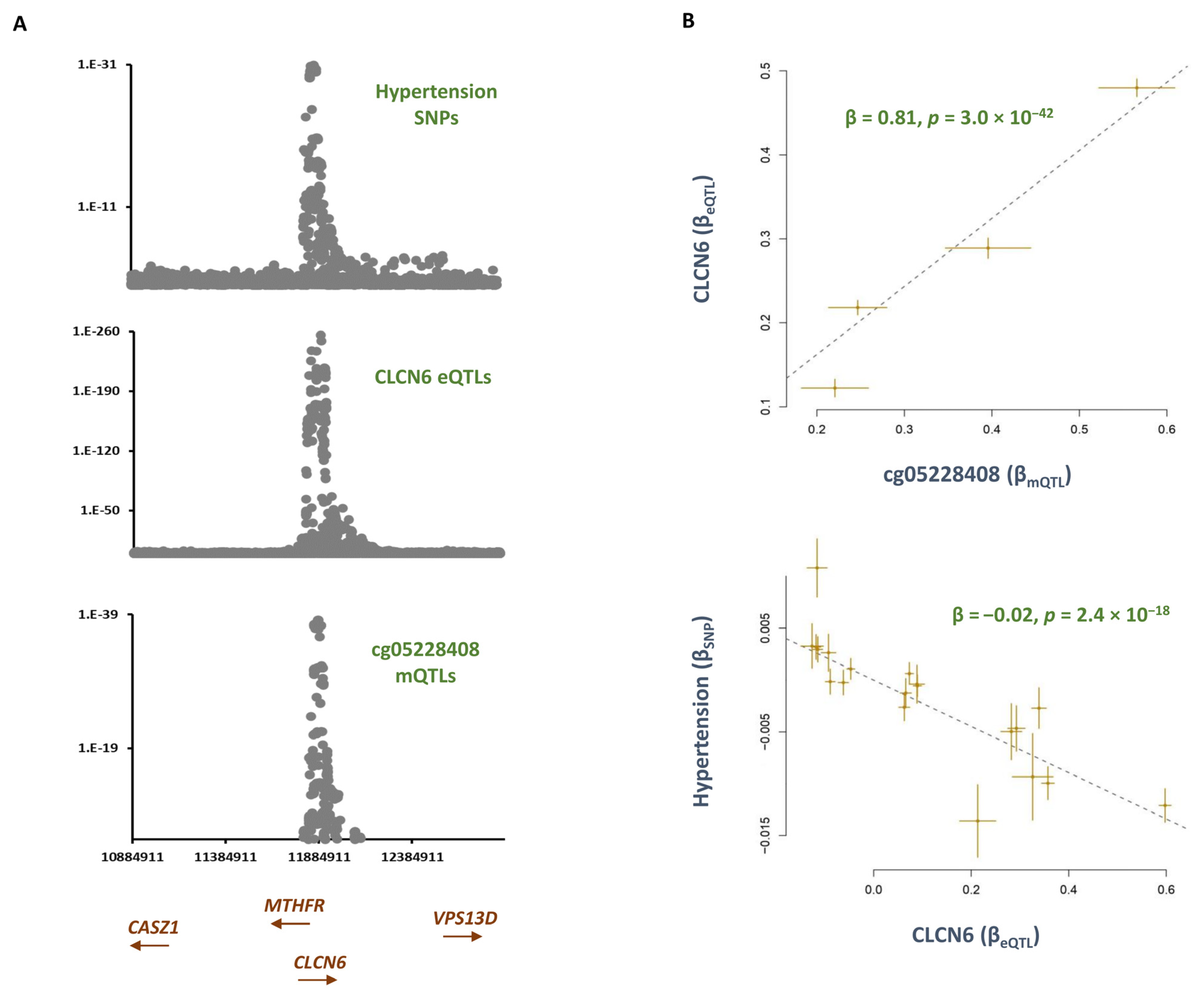
2.1. Smoking Contributes to Hypertension by Hypomethylating the cg05228408 Site and Consequently Lowering the Expression of CLCN6
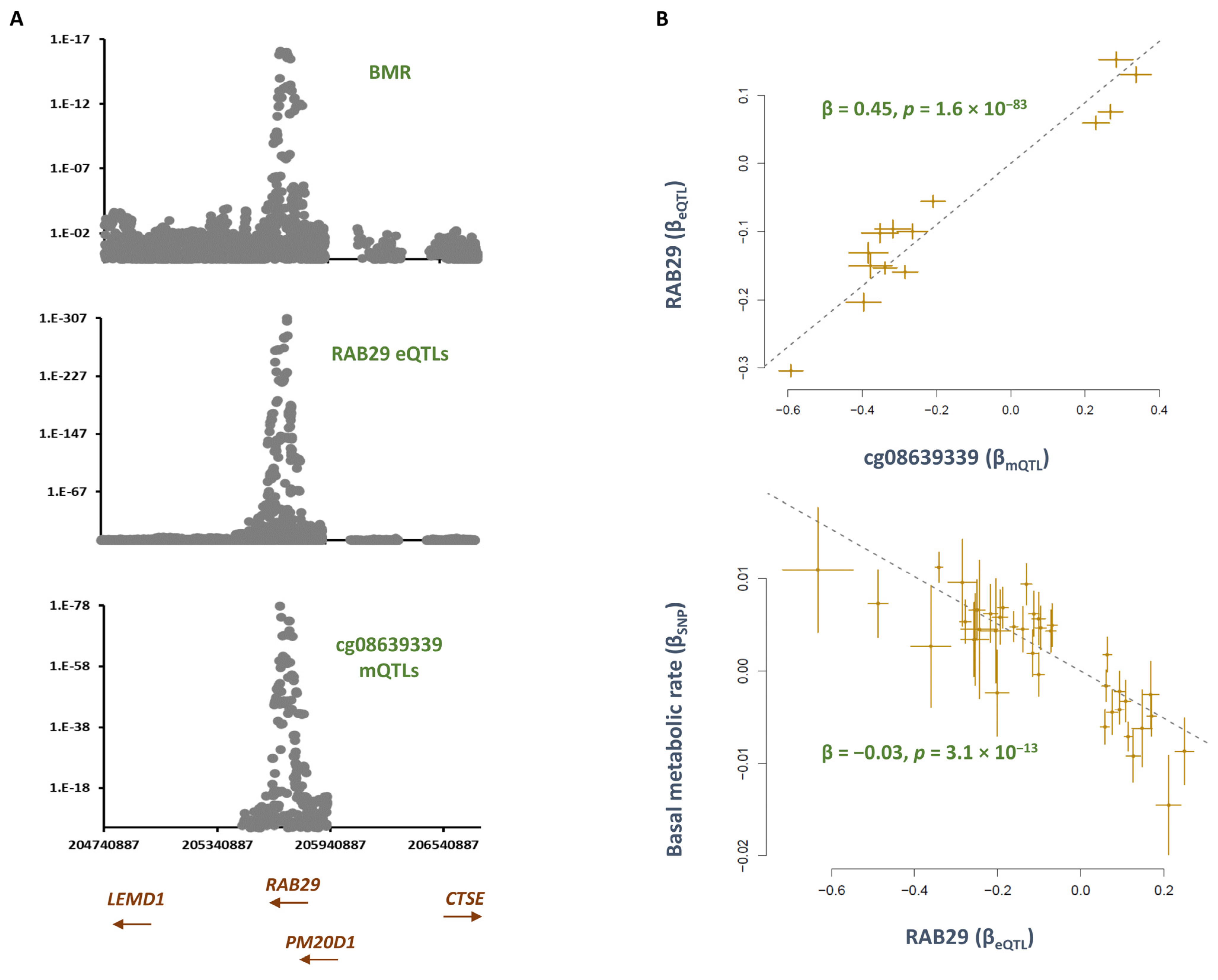
2.2. Smoking Increases the Methylation Level at cg08639339; This Lowers the Metabolic Rate by Increasing the Expression of RAB29
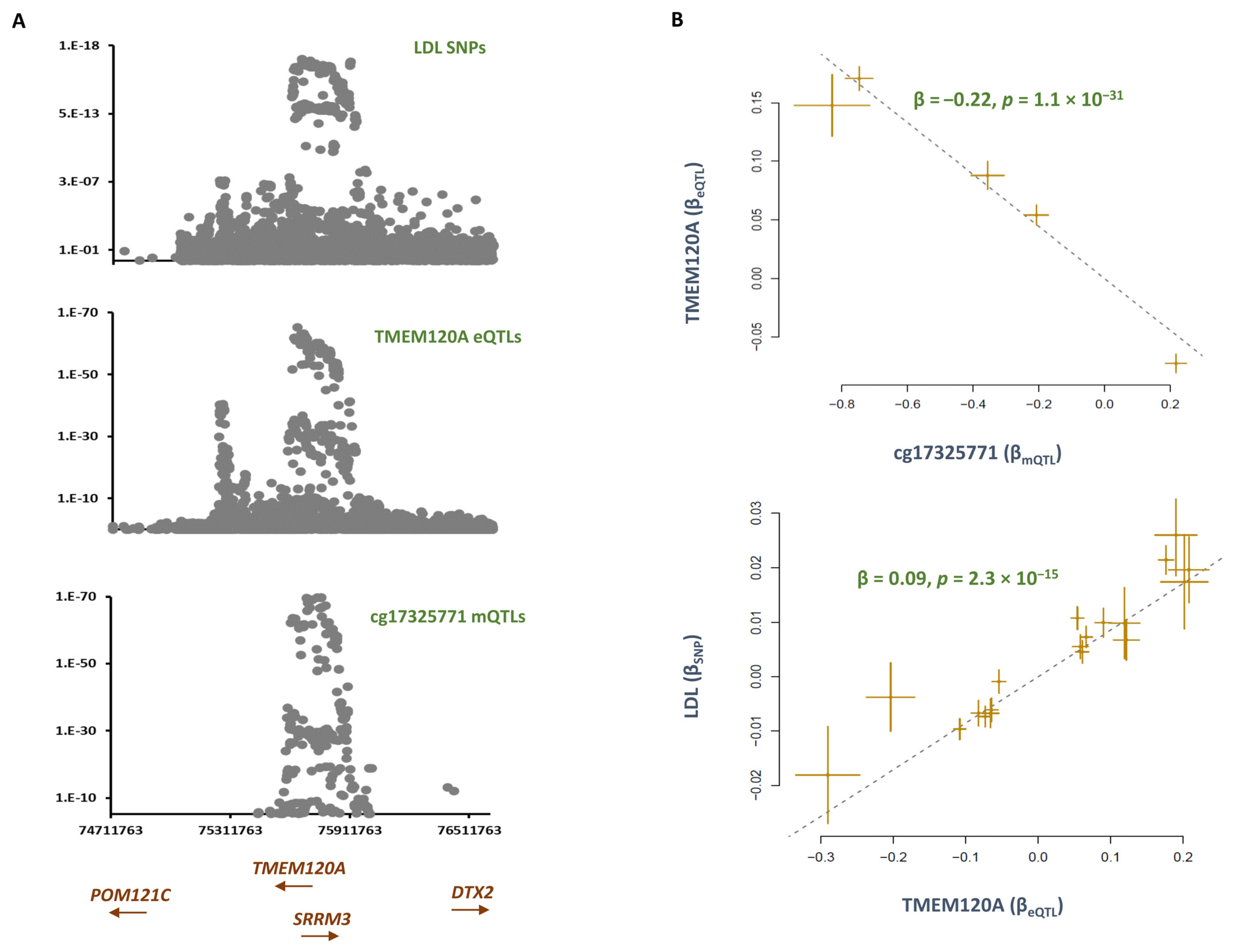
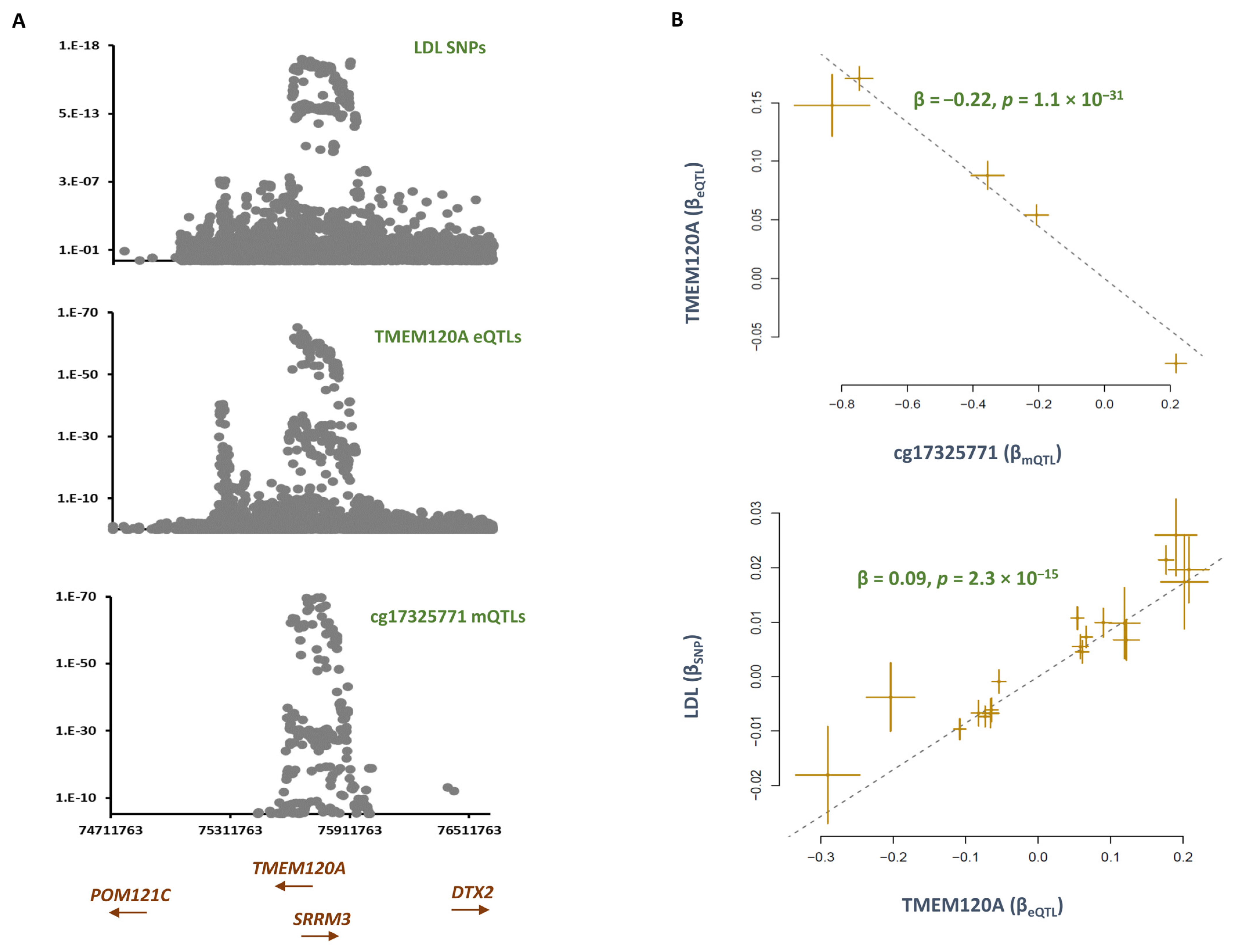
2.3. Smoking Contributes to LDL by Lowering the Methylation Level at cg17325771 and Consequently Enhancing the Expression of TMEM120A
2.4. Smoking Increases the Heart Rate by Increasing the Methylation Level at cg07029024 and Lowering the Expression of LTBP3
2.5. From Genes to Pathways
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Data Sources
5.2. Association with Cardiometabolic Traits
5.3. Pathway Analysis
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zheng, Z.; Zhang, F.; Wu, Y.; Trzaskowski, M.; Maier, R.; Robinson, M.R.; McGrath, J.J.; Visscher, P.M.; Wray, N.R.; et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 2018, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Ueda, J.; Yamazaki, T.; Funakoshi, H. Toward the Development of Epigenome Editing-Based Therapeutics: Potentials and Challenges. Int. J. Mol. Sci. 2023, 24, 4778. [Google Scholar] [CrossRef] [PubMed]
- Ligthart, S.; Steenaard, R.V.; Peters, M.J.; van Meurs, J.B.J.; Sijbrands, E.J.G.; Uitterlinden, A.G.; Bonder, M.J.; BIOS consortium; Hofman, A.; Franco, O.H.; et al. Tobacco smoking is associated with DNA methylation of diabetes susceptibility genes. Diabetologia 2016, 59, 998–1006. [Google Scholar] [CrossRef]
- Maas, S.C.E.; Mens, M.M.J.; Kühnel, B.; van Meurs, J.B.J.; Uitterlinden, A.G.; Peters, A.; Prokisch, H.; Herder, C.; Grallert, H.; Kunze, S.; et al. Smoking-related changes in DNA methylation and gene expression are associated with cardio-metabolic traits. Clin. Epigenetics 2020, 12, 157. [Google Scholar] [CrossRef]
- Fragou, D.; Pakkidi, E.; Aschner, M.; Samanidou, V.; Kovatsi, L. Smoking and DNA methylation: Correlation of methylation with smoking behavior and association with diseases and fetus development following prenatal exposure. Food Chem. Toxicol. 2019, 129, 312–327. [Google Scholar] [CrossRef]
- Joehanes, R.; Just, A.C.; Marioni, R.E.; Pilling, L.C.; Reynolds, L.M.; Mandaviya, P.R.; Guan, W.; Xu, T.; Elks, C.E.; Aslibekyan, S.; et al. Epigenetic Signatures of Cigarette Smoking. Circ. Cardiovasc. Genet. 2016, 9, 436–447. [Google Scholar] [CrossRef]
- Li, M.; Zou, D.; Li, Z.; Gao, R.; Sang, J.; Zhang, Y.; Li, R.; Xia, L.; Zhang, T.; Niu, G.; et al. EWAS Atlas: A curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 2018, 47, D983–D988. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef]
- Klemens, C.A.; Chulkov, E.G.; Wu, J.; Khan, A.H.; Levchenko, V.; Flister, M.J.; Imig, J.D.; Kriegel, A.J.; Palygin, O.; Staruschenko, A. Loss of Chloride Channel 6 (CLC-6) Affects Vascular Smooth Muscle Contractility and Arterial Stiffness via Alterations to Golgi Calcium Stores. Hypertension 2021, 77, 582–593. [Google Scholar] [CrossRef]
- Flister, M.J.; Tsaih, S.-W.; O’Meara, C.C.; Endres, B.; Hoffman, M.J.; Geurts, A.M.; Dwinell, M.R.; Lazar, J.; Jacob, H.J.; Moreno, C. Identifying multiple causative genes at a single GWAS locus. Genome Res. 2013, 23, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Feofanova, E.V.; Lim, E.; Chen, H.; Lee, M.; Liu, C.; Cupples, L.A.; Boerwinkle, E. Exome sequence association study of levels and longitudinal change of cardiovascular risk factor phenotypes in European Americans and African Americans from the Atherosclerosis Risk in Communities Study. Genet. Epidemiol. 2021, 45, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Helip-Wooley, A.; Thoene, J.G. Sucrose-induced vacuolation results in increased expression of cholesterol biosynthesis and lysosomal genes. Exp. Cell Res. 2003, 292, 89–100. [Google Scholar] [CrossRef]
- Kuwahara, T.; Inoue, K.; D’agati, V.D.; Fujimoto, T.; Eguchi, T.; Saha, S.; Wolozin, B.; Iwatsubo, T.; Abeliovich, A. LRRK2 and RAB7L1 coordinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci. Rep. 2016, 6, 29945. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Han, Y.; Baniasadi, H.; Zeng, W.; Pei, J.; Grishin, N.V.; Wang, J.; Tu, B.P.; Jiang, Y. TMEM120A is a coenzyme A-binding membrane protein with structural similarities to ELOVL fatty acid elongase. eLife 2021, 10, e71220. [Google Scholar] [CrossRef]
- Batrakou, D.G.; Heras, J.I.d.L.; Czapiewski, R.; Mouras, R.; Schirmer, E.C. TMEM120A and B: Nuclear Envelope Transmembrane Proteins Important for Adipocyte Differentiation. PLoS ONE 2015, 10, e0127712. [Google Scholar] [CrossRef]
- Czapiewski, R.; Batrakou, D.G.; Heras, J.I.d.L.; Carter, R.N.; Sivakumar, A.; Sliwinska, M.; Dixon, C.R.; Webb, S.; Lattanzi, G.; Morton, N.M.; et al. Genomic loci mispositioning in Tmem120a knockout mice yields latent lipodystrophy. Nat. Commun. 2022, 13, 1–18. [Google Scholar] [CrossRef]
- Zhou, Y.; Cashman, T.J.; Nevis, K.R.; Obregon, P.; Carney, S.A.; Liu, Y.; Gu, A.; Mosimann, C.; Sondalle, S.; Peterson, R.E.; et al. Latent TGF-β binding protein 3 identifies a second heart field in zebrafish. Nature 2011, 474, 645–648. [Google Scholar] [CrossRef]
- Guo, D.-C.; Regalado, E.S.; Pinard, A.; Chen, J.; Lee, K.; Rigelsky, C.; Zilberberg, L.; Hostetler, E.M.; Aldred, M.; Wallace, S.E.; et al. LTBP3 Pathogenic Variants Predispose Individuals to Thoracic Aortic Aneurysms and Dissections. Am. J. Hum. Genet. 2018, 102, 706–712. [Google Scholar] [CrossRef]
- Nikpay, M.; Ravati, S.; McPherson, R. Genome-wide screening identifies DNA methylation sites that regulate the blood proteome. Epigenomics 2022, 14, 837–848. [Google Scholar] [CrossRef]
- McRae, A.F.; Marioni, R.E.; Shah, S.; Yang, J.; Powell, J.E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Bowdler, L.; Painter, J.N.; et al. Identification of 55,000 Replicated DNA Methylation QTL. Sci. Rep. 2018, 8, 17605. [Google Scholar] [CrossRef] [PubMed]
- Elsworth, B.; Lyon, M.; Alexander, T.; Liu, Y.; Matthews, P.; Hallett, J.; Bates, P.; Palmer, T.; Haberland, V.; Smith, G.D. The MRC IEU OpenGWAS data infrastructure. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hannon, E.; Dempster, E.; Viana, J.; Burrage, J.; Smith, A.R.; Macdonald, R.; Clair, D.S.; Mustard, C.; Breen, G.; Therman, S.; et al. An integrated genetic-epigenetic analysis of schizophrenia: Evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol. 2016, 17, 176. [Google Scholar] [CrossRef] [PubMed]
- Hannon, E.; Gorrie-Stone, T.J.; Smart, M.C.; Burrage, J.; Hughes, A.; Bao, Y.; Kumari, M.; Schalkwyk, L.C.; Mill, J. Leveraging DNA-Methylation Quantitative-Trait Loci to Characterize the Relationship between Methylomic Variation, Gene Expression, and Complex Traits. Am. J. Hum. Genet. 2018, 103, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CpG Site | Trait | Discovery | Replication | ||||
|---|---|---|---|---|---|---|---|
| Beta | SE | p-Value | Beta | SE | p-Value | ||
| cg05228408 | Hypertension | −0.03 | 0.003 | 2.3 × 10−20 | −0.55 | 0.06 | 3.8 × 10−23 |
| cg02998240 | Low-density lipoprotein | −0.02 | 0.002 | 1.9 × 10−21 | −0.29 | 0.03 | 2.2 × 10−21 |
| cg01465596 | Systolic blood pressure | −0.03 | 0.005 | 3.3 × 10−11 | −0.53 | 0.08 | 6.0 × 10−10 |
| cg08639339 | Basal metabolic rate | −0.02 | 0.002 | 4.1 × 10−11 | −0.32 | 0.05 | 1.5 × 10−12 |
| cg27526649 | Pulse rate | −0.48 | 0.06 | 4.8 × 10−16 | −7.93 | 0.97 | 2.1 × 10−16 |
| cg10676309 | Basal metabolic rate | −0.03 | 0.005 | 8.2 × 10−12 | −0.86 | 0.14 | 3.5 × 10−10 |
| cg11105358 | Immune reaction | −0.01 | 0.001 | 3.0 × 10−10 | −0.21 | 0.03 | 3.6 × 10−10 |
| cg05789250 | Systolic blood pressure | −0.03 | 0.005 | 1.5 × 10−9 | −0.82 | 0.15 | 1.7 × 10−8 |
| cg12583553 | Basal metabolic rate | −0.02 | 0.003 | 4.8 × 10−9 | −0.31 | 0.05 | 5.7 × 10−10 |
| cg12583553 | Body fat percentage | −0.02 | 0.003 | 1.6 × 10−10 | −0.32 | 0.05 | 2.1 × 10−10 |
| cg17325771 | Low-density lipoprotein | −0.03 | 0.004 | 6.9 × 10−14 | −0.74 | 0.09 | 1.2 × 10−15 |
| cg07029024 | Pulse rate | 0.03 | 0.004 | 1.5 × 10−9 | 0.39 | 0.06 | 3.0 × 10−10 |
| Trait | Gene Indicator | MSigDB ID | Description | r | p |
|---|---|---|---|---|---|
| Basal metabolic rate | RAB29 | M1920 | Gene network contributing to metabolic disorder | 0.07 | 3 × 10−14 |
| M5017 | Regulation of immune system | 0.07 | 8 × 10−14 | ||
| Heart rate | LTBP3 | M4547 | Regulation of cell differentiation | 0.06 | 3 × 10−10 |
| M4627 | Regulation of cell proliferation | 0.06 | 6 × 10−10 | ||
| LDL | TMEM120A | M2417 | Genes targeted by PPARG and RXRA during adipogenesis | 0.06 | 3 × 10−8 |
| Hypertension | CLCN6 | M2676 | Genes up-regulated in endothelium by treatment with VEGFA | 0.1 | 2 × 10−5 |
| M38335 | Genes implicated in abnormality of central nervous system electrophysiology | 0.06 | 6 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikpay, M. Multiomics Data Analysis Identified CpG Sites That Mediate the Impact of Smoking on Cardiometabolic Traits. Epigenomes 2023, 7, 19. https://doi.org/10.3390/epigenomes7030019
Nikpay M. Multiomics Data Analysis Identified CpG Sites That Mediate the Impact of Smoking on Cardiometabolic Traits. Epigenomes. 2023; 7(3):19. https://doi.org/10.3390/epigenomes7030019
Chicago/Turabian StyleNikpay, Majid. 2023. "Multiomics Data Analysis Identified CpG Sites That Mediate the Impact of Smoking on Cardiometabolic Traits" Epigenomes 7, no. 3: 19. https://doi.org/10.3390/epigenomes7030019
APA StyleNikpay, M. (2023). Multiomics Data Analysis Identified CpG Sites That Mediate the Impact of Smoking on Cardiometabolic Traits. Epigenomes, 7(3), 19. https://doi.org/10.3390/epigenomes7030019