

The ErbB Signaling Network and Its Potential Role in Endometrial Cancer

 ,
,  , and
, and
Abstract
1. Introduction
2. Physiology of ErbB Receptors
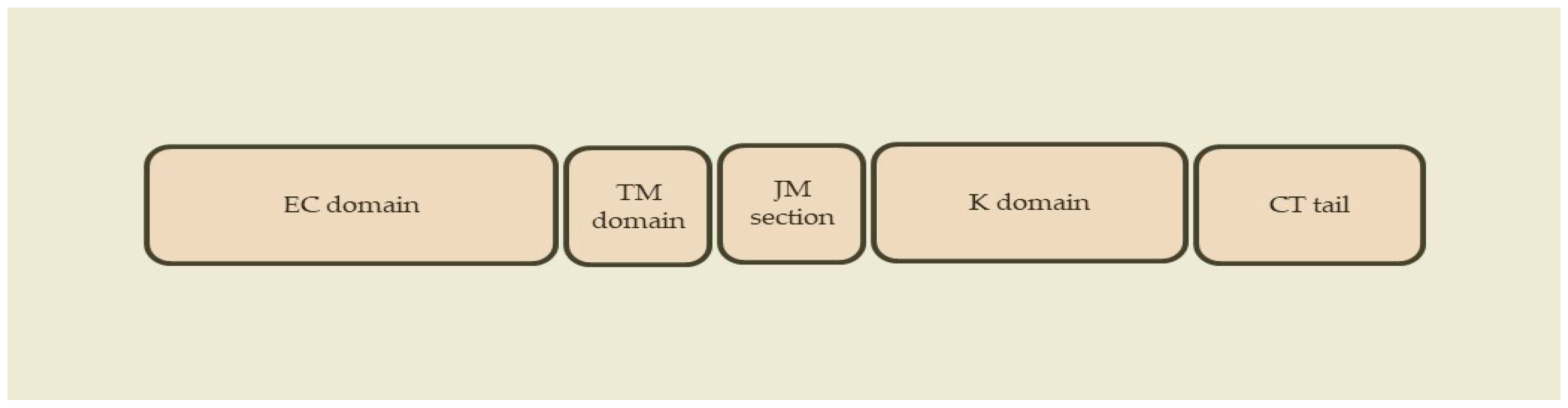
2.1. ErbB Receptors
2.2. ErbB Ligands
2.3. Receptor Homodimerization and Heterodimerization
2.4. Intracellular Tyrosine Kinase Activation
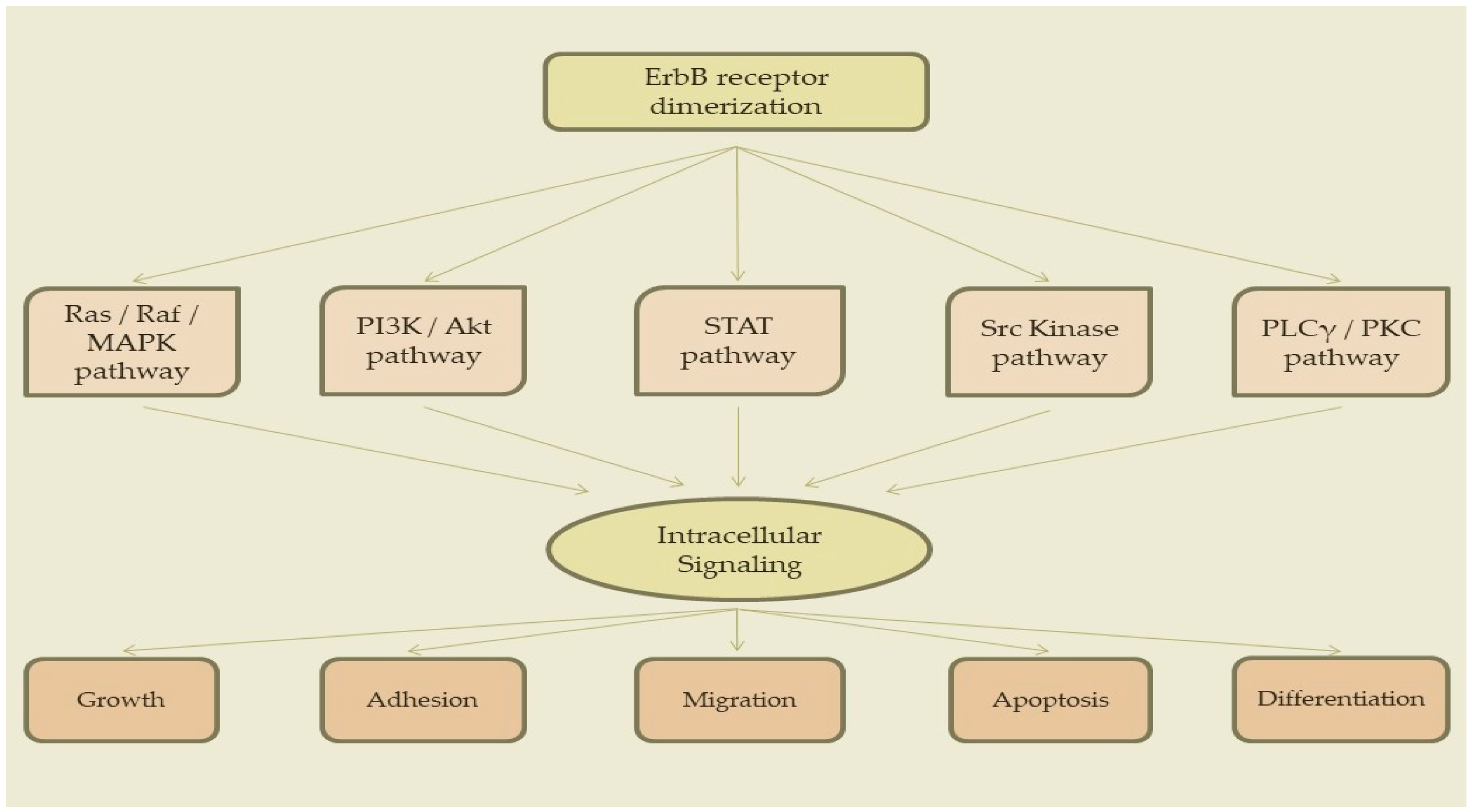
3. Signaling Pathways
3.1. Ras/Raf/MAPK Pathway
3.2. PI3K/Akt Pathway
3.3. STAT Pathway
3.4. Src Kinase Pathway
3.5. PLCγ/PKC Pathway
4. Epigenetic Regulation of ErbB Signaling
4.1. DNA Methylation
4.2. Histone Modification
4.3. Non-Coding RNAs
5. EGF Dysregulation and Carcinogenesis
5.1. Gain of Function Mutations
5.2. Genomic Amplification
5.3. Chromosomal Rearrangements
5.4. Autocrine Activation
6. ErbB Receptors in Endometrial Cancer
6.1. Profile of ErbB Receptors in Endometrial Cancer
6.2. Clinical Role in Endometrial Cancer
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- WHO Globocan. Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2020; International Agency for Research on Cancer: Lyon, France, 2020.
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Gitsch, G.; Hanzal, E.; Jensen, D.; Hacker, N.F. Endometrial cancer in premenopausal women 45 years and younger. Obstet. Gynecol. 1995, 85, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Duska, L.R.; Garrett, A.; Rueda, B.R.; Haas, J.; Chang, Y.; Fuller, A.F. Endometrial Cancer in Women 40 Years Old or Younger. Gynecol. Oncol. 2001, 83, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Erkanli, S.; Ayhan, A. Fertility-sparing therapy in young women with endometrial cancer: 2010 update. Int. J. Gynecol. Cancer 2010, 20, 1170–1187. [Google Scholar] [CrossRef] [PubMed]
- Androutsopoulos, G. Current treatment options in patients with endometrial cancer. J. Community Med. Health Educ. 2012, 2, e113. [Google Scholar] [CrossRef]
- Androutsopoulos, G.; Decavalas, G. Management of endometrial cancer. Int. J. Transl. Community Dis. 2013, 1, 101. [Google Scholar]
- Koufopoulos, N.; Carrer, D.; Koureas, N.; Sofopoulos, M.; Paraoulakis, I.; Androutsopoulos, G.; Arnogiannaki, N.; Zygouris, D.; Derdelis, G.; Terzakis, E. Pathological data on 19 cases of endometrioid carcinoma of the endometrium in women of reproductive age. Int. J. Gynecol. Cancer 2013, 23 (Suppl. 1), 322. [Google Scholar]
- Androutsopoulos, G.; Decavalas, G. Endometrial cancer: Current treatment strategies. World J. Oncol. Res. 2014, 1, 1–4. [Google Scholar]
- Androutsopoulos, G.; Michail, G.; Adonakis, G.; Decavalas, G. Current treatment approach of endometrial cancer. Int. J. Clin. Ther. Diagn. 2015, S1, 8–11. [Google Scholar]
- Androutsopoulos, G.; Adonakis, G.; Decavalas, G. Present and future in endometrial cancer treatment. Obstet. Gynecol. Int. J. 2015, 2, 00031. [Google Scholar] [CrossRef]
- ACOG. ACOG practice bulletin No. 149: Endometrial cancer. Obstet. Gynecol. 2015, 125, 1006–1026. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Creutzberg, C.; Amant, F.; Bosse, T.; Gonzalez-Martin, A.; Ledermann, J.; Marth, C.; Nout, R.; Querleu, D.; Mirza, M.R.; et al. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: Diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, 16–41. [Google Scholar] [CrossRef] [PubMed]
- Androutsopoulos, G.; Michail, G.; Decavalas, G. New insights in endometrial cancer treatment. Clin. Oncol. Endometrial Cancer 2016, 1, 1040. [Google Scholar]
- Androutsopoulos, G.; Decavalas, G. Standard and novel therapies in endometrial cancer. J. Gynecol. Women’s Health 2016, 1, 555564. [Google Scholar] [CrossRef]
- Androutsopoulos, G.; Kotsopoulos, I.; Decavalas, G. Fertility preservation in young patients with endometrial cancer. World J. Oncol. Res. 2016, 3, 36–39. [Google Scholar] [CrossRef]
- Sundar, S.; Balega, J.; Crosbie, E.; Drake, A.; Edmondson, R.; Fotopoulou, C.; Gallos, I.; Ganesan, R.; Gupta, J.; Johnson, N.; et al. BGCS uterine cancer guidelines: Recommendations for practice. Eur. J. Obstet. Gynecol. Reprod. Biol. 2017, 213, 71–97. [Google Scholar] [CrossRef]
- Androutsopoulos, G.; Kotsopoulos, I.; Korompelis, P.; Michail, G.; Adonakis, G.; Decavalas, G. Systematic lymphadenectomy or sentinel lymph node dissection in endometrial cancer: A clinical dilemma. Hell. J. Obst. Gynecol. 2017, 16, 14–19. [Google Scholar]
- Androutsopoulos, G.; Kotsopoulos, I.; Adonakis, G.; Decavalas, G. Conservative management of young patients with early stage endometrial cancer. J. Gynecol. Women’s Health 2017, 2, 555586. [Google Scholar] [CrossRef]
- Androutsopoulos, G.; Adonakis, G.; Decavalas, G. ErbB targeted therapy in endometrial cancer. In Endometrial Cancer: Current Epidemiology, Detection and Management; Farghaly, S., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2014. [Google Scholar]
- Androutsopoulos, G.; Kotsopoulos, I.; Korompelis, P.; Michail, G.; Adonakis, G.; Decavalas, G. Conservative therapeutic approach in young patients with endometrial cancer: Is it really possible? Hell. J. Obst. Gynecol. 2017, 16, 7–23. [Google Scholar]
- Michail, G.D. Endometrial Cancer-Diagnosis. Int. J. Clin. Ther. Diagn. 2015, 1, 17–27. [Google Scholar]
- Weimer, J.; Hüttmann, M.; Nusilati, A.; Andreas, S.; Röseler, J.; Tribian, N.; Rogmans, C.; Stope, M.B.; Dahl, E.; Mustea, A.; et al. Fluorescence in situ hybridization test for detection of endometrial carcinoma cells by non-invasive vaginal swab. J. Cell. Mol. Med. 2023, 27, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Pouliakis, A.; Damaskou, V.; Margari, N.; Karakitsou, E.; Pergialiotis, V.; Valasoulis, G.; Michail, G.; Chrelias, C.; Chrelias, G.; Sioulas, V.; et al. Artificial Intelligence and Image Analysis for the Identification of Endometrial Malignancies: A Comparative Study. In Research Anthology on Medical Informatics in Breast and Cervical Cancer; I.R. Management Association, Ed.; IGI Global: Hershey, PA, USA, 2023; pp. 1–30. [Google Scholar]
- Pouliakis, A.; Damaskou, V.; Margari, N.; Karakitsou, E.; Pergialiotis, V.; Valasoulis, G.; Michail, G.; Chrelias, C.; Chrelias, G.; Sioulas, V.; et al. Artificial Intelligence and Image Analysis for the Identification of Endometrial Malignancies: A Comparative Study. In Quality Assurance in the Era of Individualized Medicine; Moumtzoglou, A.S., Ed.; IGI Global: Hershey, PA, USA, 2020; pp. 110–146. [Google Scholar]
- Pouliakis, A.; Margari, N.; Karakitsou, E.; Valasoulis, G.; Koufopoulos, N.; Koureas, N.; Alamanou, E.; Pergialiotis, V.; Damaskou, V.; Panayiotides, I.G. Artificial Intelligence via Competitive Learning and Image Analysis for Endometrial Malignancies: Discriminating Endometrial Cells and Lesions. Int. J. Reliab. Qual. E Healthc. 2019, 8, 38–54. [Google Scholar] [CrossRef]
- Piedimonte, S.; Rosa, G.; Gerstl, B.; Sopocado, M.; Coronel, A.; Lleno, S.; Vicus, D. Evaluating the use of machine learning in endometrial cancer: A systematic review. Int. J. Gynecol. Cancer 2023, 33, 1383–1393. [Google Scholar] [CrossRef]
- Purandare, N.C.; Trevisan, J.; Patel, I.I.; Gajjar, K.; Mitchell, A.L.; Theophilou, G.; Valasoulis, G.; Martin, M.; von Bünau, G.; Kyrgiou, M.; et al. Exploiting biospectroscopy as a novel screening tool for cervical cancer: Towards a framework to validate its accuracy in a routine clinical setting. Bioanalysis 2013, 5, 2697–2711. [Google Scholar] [CrossRef]
- Theophilou, G.; Morais, C.L.M.; Halliwell, D.E.; Lima, K.M.G.; Drury, J.; Martin-Hirsch, P.L.; Stringfellow, H.F.; Hapangama, D.K.; Martin, F.L. Synchrotron- and focal plane array-based Fourier-transform infrared spectroscopy differentiates the basalis and functionalis epithelial endometrial regions and identifies putative stem cell regions of human endometrial glands. Anal. Bioanal. Chem. 2018, 410, 4541–4554. [Google Scholar] [CrossRef]
- Jacobs, I.; Gentry-Maharaj, A.; Burnell, M.; Manchanda, R.; Singh, N.; Sharma, A.; Ryan, A.; Seif, M.W.; Amso, N.N.; Turner, G.; et al. Sensitivity of transvaginal ultrasound screening for endometrial cancer in postmenopausal women: A case-control study within the UKCTOCS cohort. Lancet Oncol. 2010, 12, 38–48. [Google Scholar] [CrossRef]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Doll, A.; Abal, M.; Rigau, M.; Monge, M.; Gonzalez, M.; Demajo, S.; Colás, E.; Llauradó, M.; Alazzouzi, H.; Planagumá, J.; et al. Novel molecular profiles of endometrial cancer-new light through old windows. J. Steroid. Biochem. Mol. Biol. 2008, 108, 221–229. [Google Scholar] [CrossRef]
- Kandoth, C.; Schultz, N.; Cherniack, A.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.; Pashtan, I.; Shen, R.; Benz, C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Talhouk, A.; McConechy, M.; Leung, S.; Yang, W.; Lum, A.; Senz, J.; Boyd, N.; Pike, J.; Anglesio, M.; Kwon, J.; et al. Confirmation of ProMisE: A simple, genomics-based clinical classifier for endometrial cancer. Cancer 2017, 123, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Le Gallo, M.; Bell, D. The emerging genomic landscape of endometrial cancer. Clin. Chem. 2014, 60, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Holbro, T.; Civenni, G.; Hynes, N.E. The ErbB receptors and their role in cancer progression. Exp. Cell Res. 2003, 284, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.D.; Skaria, K.B.; Yarden, Y. Signal transduction and oncogenesis by ErbB/HER receptors. Int. J. Radiat. Oncol. 2004, 58, 903–913. [Google Scholar] [CrossRef]
- Überall, I.; Kolář, Z.; Trojanec, R.; Berkovcová, J.; Hajdúch, M. The status and role of ErbB receptors in human cancer. Exp. Mol. Pathol. 2008, 84, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- McDonell, L.M.; Kernohan, K.D.; Boycott, K.M.; Sawyer, S.L. Receptor tyrosine kinase mutations in developmental syndromes and cancer: Two sides of the same coin. Hum. Mol. Genet. 2015, 24, R60–R66. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Casalini, P.; Iorio, M.V.; Galmozzi, E.; Ménard, S. Role of HER receptors family in development and differentiation. J. Cell. Physiol. 2004, 200, 343–350. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef]
- Linggi, B.; Carpenter, G. ErbB receptors: New insights on mechanisms and biology. Trends Cell Biol. 2006, 16, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. The Croonian Lecture 1997. The phosphorylation of proteins on tyrosine: Its role in cell growth and disease. Philos. Trans. R. Soc. B Biol. Sci. 1998, 353, 583–605. [Google Scholar] [CrossRef] [PubMed]
- Mass, R.D. The HER receptor family: A rich target for therapeutic development. Int. J. Radiat. Oncol. 2004, 58, 932–940. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.-S.; Leahy, D.J.; Lemmon, M.A. EGF Activates Its Receptor by Removing Interactions that Autoinhibit Ectodomain Dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Dawson, J.P.; Berger, M.; Lin, C.-C.; Schlessinger, J.; Lemmon, M.A.; Ferguson, K.M. Epidermal Growth Factor Receptor Dimerization and Activation Require Ligand-Induced Conformational Changes in the Dimer Interface. Mol. Cell. Biol. 2005, 25, 7734–7742. [Google Scholar] [CrossRef]
- Özcan, F.; Klein, P.; Lemmon, M.A.; Lax, I.; Schlessinger, J. On the nature of low- and high-affinity EGF receptors on living cells. Proc. Natl. Acad. Sci. USA 2006, 103, 5735–5740. [Google Scholar] [CrossRef]
- Olayioye, M.; Neve, R.; Lane, H.; Hynes, N. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef]
- Qian, X.; LeVea, C.; Freeman, J.; Dougall, W.; Greene, M. Heterodimerization of epidermal growth factor receptor and wild-type or kinase-deficient Neu: A mechanism of interreceptor kinase activation and transphosphorylation. Proc. Natl. Acad. Sci. USA 1994, 91, 1500–1504. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The Crystal Structure of a Truncated ErbB2 Ectodomain Reveals an Active Conformation, Poised to Interact with Other ErbB Receptors. Mol. Cell 2003, 11, 495–505. [Google Scholar] [CrossRef]
- Citri, A.; Skaria, K.B.; Yarden, Y. The deaf and the dumb: The biology of ErbB-2 and ErbB-3. Exp. Cell Res. 2003, 284, 54–65. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar]
- Zhou, S.; Shoelson, S.E.; Chaudhuri, M.; Gish, G.; Pawson, T.; Haser, W.G.; King, F.; Roberts, T.; Ratnofsky, S.; Lechleider, R.J.; et al. SH2 domains recognize specific phosphopeptide sequences. Cell 1993, 72, 767–778. [Google Scholar] [CrossRef]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer therapies. J. Clin. Investig. 2007, 117, 2051–2058. [Google Scholar] [CrossRef]
- Scaltriti, M.; Baselga, J. The Epidermal Growth Factor Receptor Pathway: A Model for Targeted Therapy. Clin. Cancer Res. 2006, 12, 5268–5272. [Google Scholar] [CrossRef]
- Molina, J.; Adjei, A. The Ras/Raf/MAPK pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef]
- Cary, L.; Han, D.; Guan, J. Integrin-mediated signal transduction pathways. Histol. Histopathol. 1999, 14, 1001–1009. [Google Scholar] [PubMed]
- Stacey, D.W. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr. Opin. Cell Biol. 2003, 15, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, O.; Gebbink, M.F.; Voest, E.E. Stimulation of angiogenesis by Ras proteins. Biochim. Biophys. Acta Rev. Cancer 2004, 1654, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, E.; Daly, R.; Batzer, A.; Li, W.; Margolis, B.; Lammers, R.; Ullrich, A.; Skolnik, E.; Bar-Sagi, D.; Schlessinger, J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992, 70, 431–442. [Google Scholar] [CrossRef]
- Rozakis-Adcock, M.; McGlade, J.; Mbamalu, G.; Pelicci, G.; Daly, R.; Li, W.; Batzer, A.; Thomas, S.; Brugge, J.; Pelicci, P.G.; et al. Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature 1992, 360, 689–692. [Google Scholar] [CrossRef]
- Batzer, A.G.; Rotin, D.; Urena, J.M.; Skolnik, E.Y.; Schlessinger, J. Hierarchy of Binding Sites for Grb2 and Shc on the Epidermal Growth Factor Receptor. Mol. Cell. Biol. 1994, 14, 5192–5201. [Google Scholar]
- Buday, L. Membrane-targeting of signalling molecules by SH2/SH3 domain-containing adaptor proteins. Biochim. Biophys. Acta 1999, 1422, 187–204. [Google Scholar] [CrossRef]
- Guo, Y.; Pan, W.; Liu, S.B.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Pierre, S.; Bats, A.-S.; Coumoul, X. Understanding SOS (Son of Sevenless). Biochem. Pharmacol. 2011, 82, 1049–1056. [Google Scholar] [CrossRef]
- Bandaru, P.; Kondo, Y.; Kuriyan, J. The Interdependent Activation of Son-of-Sevenless and Ras. Cold Spring Harb. Perspect. Med. 2018, 9, a031534. [Google Scholar] [CrossRef]
- Freedman, T.; Sondermann, H.; Friedland, G.; Kortemme, T.; Bar-Sagi, D.; Marqusee, S.; Kuriyan, J. A Ras-induced conformational switch in the Ras activator Son of sevenless. Proc. Natl. Acad. Sci. USA 2006, 103, 16692–16697. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Marais, R.; Light, Y.; Paterson, H.; Marshall, C. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995, 14, 3136–3145. [Google Scholar] [CrossRef] [PubMed]
- Stokoe, D.; McCormick, F. Activation of c-Raf-1 by Ras and Src through different mechanisms: Activation in vivo and in vitro. EMBO J. 1997, 16, 2384–2396. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Hemmings, B.; Restuccia, D. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Alessi, D. The PI3K-PDK1 connection: More than just a road to PKB. Biochem. J. 2000, 346, 561–576. [Google Scholar] [CrossRef]
- Stokoe, D.; Stephens, L.R.; Copeland, T.; Gaffney, P.R.J.; Reese, C.B.; Painter, G.F.; Holmes, A.B.; McCormick, F.; Hawkins, P.T. Dual Role of Phosphatidylinositol-3,4,5-trisphosphate in the Activation of Protein Kinase B. Science 1997, 277, 567–570. [Google Scholar] [CrossRef]
- Carpenter, C.; Auger, K.; Chanudhuri, M.; Yoakim, M.; Schaffhausen, B.; Shoelson, S.; Cantley, L. Phosphoinositide 3-kinase is activated by phosphopeptides that bind to the SH2 domains of the 85-kDa subunit. J. Biol. Chem. 1993, 268, 9478–9483. [Google Scholar] [CrossRef] [PubMed]
- Mattoon, D.R.; Lamothe, B.; Lax, I.; Schlessinger, J. The docking protein Gab1 is the primary mediator of EGF-stimulated activation of the PI-3K/Akt cell survival pathway. BMC Biol. 2004, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, J.; Rosler, K.; Harrison, D. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Haura, E.B.; Turkson, J.; Jove, R. Mechanisms of Disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat. Clin. Pract. Oncol. 2005, 2, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D. The Jak/STAT pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011205. [Google Scholar] [CrossRef] [PubMed]
- Olayioye, M.A.; Beuvink, I.; Horsch, K.; Daly, J.M.; Hynes, N.E. ErbB Receptor-induced Activation of Stat Transcription Factors Is Mediated by Src Tyrosine Kinases. J. Biol. Chem. 1999, 274, 17209–17218. [Google Scholar] [CrossRef]
- Haura, E. SRC and STAT pathways. J. Thorac. Oncol. 2006, 1, 403–405. [Google Scholar] [CrossRef]
- Playford, M.P.; Schaller, M.D. The interplay between Src and integrins in normal and tumor biology. Oncogene 2004, 23, 7928–7946. [Google Scholar] [CrossRef]
- Bromann, P.; Korkaya, H.; Courtneidge, S. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene 2004, 23, 7957–7968. [Google Scholar] [CrossRef]
- Leu, T.; Maa, M. Functional implication of the interaction between EGF receptor and c-Src. Front. Biosci. 2003, 8, s28–s38. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Frame, M. Src in cancer: Deregulation and consequences for cell behaviour. Biochim. Biophys. Acta 2002, 1602, 114–130. [Google Scholar] [CrossRef] [PubMed]
- Yeatman, T. A renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.; Carpenter, G. Phospholipase C-gamma1: Regulation of enzyme function and role in growth factor-dependent signal transduction. Cytokine Growth Factor Rev. 1997, 8, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Rotin, D.; Honegger, A.; Margolis, B.; Ullrich, A.; Schlessinger, J. Presence of SH2 domains of phospholipase C gamma 1 enhances substrate phosphorylation by increasing the affinity toward the epidermal growth factor receptor. J. Biol. Chem. 1992, 267, 9678–9683. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; Vecchi, M.; Ji, Q.; Mernaugh, R.; Carpenter, G. The role of individual SH2 domains in mediating association of phospholipase C-gamma1 with the activated EGF receptor. J. Biol. Chem. 1999, 274, 26091–26097. [Google Scholar] [CrossRef]
- Kadamur, G.; Ross, E. Mammalian phospholipase C. Annu. Rev. Physiol. 2013, 75, 127–154. [Google Scholar] [CrossRef]
- Marais, R.; Light, Y.; Mason, C.; Paterson, H.; Olson, M.F.; Marshall, C.J. Requirement of Ras-GTP-Raf Complexes for Activation of Raf-1 by Protein Kinase C. Science 1998, 280, 109–112. [Google Scholar] [CrossRef]
- Schönwasser, D.C.; Marais, R.M.; Marshall, C.J.; Parker, P.J. Activation of the Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Pathway by Conventional, Novel, and Atypical Protein Kinase C Isotypes. Mol. Cell. Biol. 1998, 18, 790–798. [Google Scholar] [CrossRef]
- Spangle, J.M.; Roberts, T.M. Epigenetic regulation of RTK signaling. J. Mol. Med. 2017, 95, 791–798. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics: Figure 1. Minerva Anestesiol. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Inoue, F.; Sone, K.; Toyohara, Y.; Takahashi, Y.; Kukita, A.; Hara, A.; Taguchi, A.; Tanikawa, M.; Tsuruga, T.; Osuga, Y. Targeting Epigenetic Regulators for Endometrial Cancer Therapy: Its Molecular Biology and Potential Clinical Applications. Int. J. Mol. Sci. 2021, 22, 2305. [Google Scholar] [CrossRef] [PubMed]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA Methylation in Cancer and Aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef] [PubMed]
- Retis-Resendiz, A.M.; González-García, I.N.; León-Juárez, M.; Camacho-Arroyo, I.; Cerbón, M.; Vázquez-Martínez, E.R. The role of epigenetic mechanisms in the regulation of gene expression in the cyclical endometrium. Clin. Epigenetics 2021, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Lee, H.; Kim, W. Promoter methylation and silencing of PTEN in gastric carcinoma. Lab. Investig. 2002, 82, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Song, H.; Li, C.; Labaff, A.; Lim, S.; Li, L.; Kan, S.; Chen, Y.; Zhang, K.; Lang, J.; Xie, X.; et al. Acetylation of EGF receptor contributes to tumor cell resistance to histone deacetylase inhibitors. Biochem. Biophys. Res. Commun. 2011, 404, 68–73. [Google Scholar] [CrossRef]
- Wei, J.; Huang, K.; Yang, C.; Kang, C. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 2017, 373–379. [Google Scholar]
- Pathania, A.S. Crosstalk between Noncoding RNAs and the Epigenetics Machinery in Pediatric Tumors and Their Microenvironment. Cancers 2023, 15, 2833. [Google Scholar] [CrossRef]
- Kumar, S.; Gonzalez, E.A.; Rameshwar, P.; Etchegaray, J.-P. Non-Coding RNAs as Mediators of Epigenetic Changes in Malignancies. Cancers 2020, 12, 3657. [Google Scholar] [CrossRef]
- Schickel, R.; Boyerinas, B.; Park, S.; Peter, M. MicroRNAs: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 2008, 27, 5959–5974. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Maizel, A.; Chen, X. Traffic into silence: Endomembranes and post-transcriptional RNA silencing. EMBO J. 2014, 33, 968–980. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Clark, M.B.; Mattick, J.S. Long noncoding RNAs in cell biology. Semin. Cell Dev. Biol. 2011, 22, 366–376. [Google Scholar]
- Morlando, M.; Fatica, A. Alteration of Epigenetic Regulation by Long Noncoding RNAs in Cancer. Int. J. Mol. Sci. 2018, 19, 570. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Agarwal, S.; Johnson, J.L.; Galia, L.; Lei, X.; Stein, K.; Olagnier, D.; Gaede, K.I.; Herzmann, C.; Holm, C.K.; et al. The lncRNA LUCAT1 is elevated in inflammatory disease and restrains inflammation by regulating the splicing and stability of NR4A2. Proc. Natl. Acad. Sci. USA 2023, 120, e2213715120. [Google Scholar] [CrossRef]
- Sauvageau, M. Diverging RNPs: Toward Understanding lncRNA-Protein Interactions and Functions. Adv. Exp. Med. Biol. 2019, 1203, 285–312. [Google Scholar]
- Ninomiya, K.; Adachi, S.; Natsume, T.; Iwakiri, J.; Terai, G.; Asai, K.; Hirose, T. LncRNA-dependent nuclear stress bodies promote intron retention through SR protein phosphorylation. EMBO J. 2020, 39, e102729. [Google Scholar] [CrossRef]
- Taniue, K.; Kurimoto, A.; Sugimasa, H.; Nasu, E.; Takeda, Y.; Iwasaki, K.; Nagashima, T.; Okada-Hatakeyama, M.; Oyama, M.; Kozuka-Hata, H.; et al. Long noncoding RNA UPAT promotes colon tumorigenesis by inhibiting degradation of UHRF1. Proc. Natl. Acad. Sci. USA 2016, 113, 1273–1278. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Li, X.; Yi, S.; Xu, J. Gain-of-Function Mutations: An emerging advantage for cancer biology. Trends Biochem. Sci. 2019, 44, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.; Campbell, P.; Futreal, P. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Pon, J.; Marra, M. Driver and passenger mutations in cancer. Annu. Rev. Pathol. 2015, 10, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Albertson, D.; Collins, C.; McCormick, F.; Gray, J. Chromosome aberrations in solid tumors. Nat. Genet. 2003, 34, 369–376. [Google Scholar] [CrossRef]
- Albertson, D. Gene amplification in cancer. Trends Genet. 2006, 22, 447–455. [Google Scholar] [CrossRef]
- Jen, K. Gene Amplification. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 171–172. [Google Scholar]
- Worthylake, R.; Opresko, L.K.; Wiley, H.S. ErbB-2 Amplification Inhibits Down-regulation and Induces Constitutive Activation of Both ErbB-2 and Epidermal Growth Factor Receptors. J. Biol. Chem. 1999, 274, 8865–8874. [Google Scholar] [CrossRef]
- Carraway, K., 3rd; Sweeney, C. EGF receptor activation by heterologous mechanisms. Cancer Cell 2002, 1, 405–406. [Google Scholar] [CrossRef]
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 2012, 18, 1630–1638. [Google Scholar] [CrossRef]
- Hasty, P.; Montagna, C. Chromosomal Rearrangements in Cancer: Detection and potential causal mechanisms. Mol. Cell. Oncol. 2014, 1, e29904. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef]
- Sporn, M.; Todaro, G. Autocrine secretion and malignant transformation of cells. N. Engl. J. Med. 1980, 303, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.; Karnes, W.; Cuttitta, F.; Walker, A. Autocrine growth factors and solid tumor malignancy. West. J. Med. 1991, 155, 152–163. [Google Scholar]
- Rizzino, A. Understanding the roles of growth factors in carcinogenesis: Modulation of autocrine growth control by differentiation. Int. J. Dev. Biol. 1993, 37, 61–65. [Google Scholar]
- Nicholson, K.; Streuli, C.; Anderson, N. Autocrine Signalling Through erbB Receptors Promotes Constitutive Activation of Protein Kinase B/Akt in Breast Cancer Cell Lines. Breast Cancer Res. Treat. 2003, 81, 117–128. [Google Scholar] [CrossRef]
- Singh, A.B.; Harris, R.C. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell. Signal. 2005, 17, 1183–1193. [Google Scholar] [CrossRef]
- Srinivasan, R.; Benton, E.; McCormick, F.; Thomas, H.; Gullick, W. Expression of the c-erbB-3/HER-3 and c-erbB-4/HER-4 growth factor receptors and their ligands, neuregulin-1 alpha, neuregulin-1 beta, and betacellulin, in normal endometrium and endometrial cancer. Clin. Cancer Res. 1999, 5, 2877–2883. [Google Scholar]
- Ejskjaer, K.; Sørensen, B.; Poulsen, S.; Mogensen, O.; Forman, A.; Nexø, E. Expression of the epidermal growth factor system in human endometrium during the menstrual cycle. Mol. Hum. Reprod. 2005, 11, 543–551. [Google Scholar] [CrossRef]
- Ejskjaer, K.; Sorensen, B.S.; Poulsen, S.S.; Forman, A.; Nexo, E.; Mogensen, O. Expression of the epidermal growth factor system in endometrioid endometrial cancer. Gynecol. Oncol. 2007, 104, 158–167. [Google Scholar] [CrossRef]
- Brys, M.; Semczuk, A.; Rechberger, T.; Krajewska, W.M. Expression of erbB-1 and erbB-2 genes in normal and pathological human endometrium. Oncol. Rep. 2007, 18, 261–265. [Google Scholar] [CrossRef][Green Version]
- Reinartz, J.; George, E.; Lindgren, B.; Niehans, G. Expression of p53, transforming growth factor alpha, epidermal growth factor receptor, and c-erbB-2 in endometrial carcinoma and correlation with survival and known predictors of survival. Hum. Pathol. 1994, 25, 1075–1083. [Google Scholar] [CrossRef]
- Khalifa, M.A.; Mannel, R.S.; Haraway, S.D.; Walker, J.; Min, K.-W. Expression of EGFR, HER-2/neu, P53, and PCNA in Endometrioid, Serous Papillary, and Clear Cell Endometrial Adenocarcinomas. Gynecol. Oncol. 1994, 53, 84–92. [Google Scholar] [CrossRef]
- Scambia, G.; Panici, P.B.; Ferrandina, G.; Battaglia, F.; Distefano, M.; D'Andrea, G.; De Vincenzo, R.; Maneschi, F.; Ranelletti, F.O.; Mancuso, S. Significance of epidermal growth factor receptor expression in primary human endometrial cancer. Int. J. Cancer 2007, 56, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Niikura, H.; Sasano, H.; Kaga, K.; Sato, S.; Yajima, A. Expression of epidermal growth factor family proteins and epidermal growth factor receptor in human endometrium. Hum. Pathol. 1996, 27, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Santos, L.; Winterhoff, B.; Hatmal, M.; Keeney, G.L.; Mariani, A.; Jones, M.; Neuper, C.; Thomas, B.; Muderspach, L.; et al. HER2 gene amplification and EGFR expression in a large cohort of surgically staged patients with nonendometrioid (type II) endometrial cancer. Br. J. Cancer 2008, 100, 89–95. [Google Scholar] [CrossRef]
- Adonakis, G.; Androutsopoulos, G.; Koumoundourou, D.; Liava, A.; Ravazoula, P.; Kourounis, G. Expression of the epidermal growth factor system in endometrial cancer. Eur. J. Gynaecol. Oncol. 2008, 29, 450–454. [Google Scholar]
- Adonakis, G.; Androutsopoulos, G. The role of ErbB receptors in endometrial cancer. In Cancer of the Uterine Endometrium—Advances and Controversies; Saldivar, J.S., Ed.; IntechOpen: London, UK, 2012; pp. 23–38. [Google Scholar]
- Androutsopoulos, G.; Adonakis, G.; Gkermpesi, M.; Gkogkos, P.; Ravazoula, P.; Kourounis, G. Expression of the epidermal growth factor system in endometrial cancer after adjuvant tamoxifen treatment for breast cancer. Eur. J. Gynaecol. Oncol. 2006, 27, 490–494. [Google Scholar]
- Reyes, H.D.; Thiel, K.W.; Carlson, M.J.; Meng, X.; Yang, S.; Stephan, J.-M.; Leslie, K.K. Comprehensive profiling of EGFR/HER receptors for personalized treatment of gynecologic cancers. Mol. Diagn. Ther. 2014, 18, 137–151. [Google Scholar] [CrossRef]
- Androutsopoulos, G.; Michail, G.; Adonakis, G.; Decavalas, G. Molecular mechanisms, expression and clinical role of ErbB receptors in endometrial cancer. Int. J. Clin. Ther. Diagn. 2015, S1, 28–32. [Google Scholar]
- Michail, G.; Styliara, I.; Panas, P.; Markatos, F.; Koumoundourou, D.; Ravazoula, P.; Adonakis, G.; Androutsopoulos, G. EP472 ErbB receptors profile in non-selected patients with endometrial cancer. Int. J. Gynecol. Cancer 2019, 29, A298–A299. [Google Scholar]
- Styliara, I.; Zarogianni, E.; Panas, P.; Michail, G.; Koumoundourou, D.; Ravazoula, P.; Adonakis, G.; Androutsopoulos, G. 299 EGF System receptors profiling in various histologic subgroups of endometrial cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 2022, 270, e85–e86. [Google Scholar] [CrossRef]
- Androutsopoulos, G.; Adonakis, G.; Liava, A.; Ravazoula, P.; Decavalas, G. Expression and potential role of ErbB receptors in type II endometrial cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 168, 204–208. [Google Scholar] [CrossRef]
- Michail, G.; Panas, P.; Markatos, F.; Styliara, I.; Koumoundourou, D.; Ravazoula, P.; Adonakis, G.; Androutsopoulos, G. ErbB receptors profiling in selected patients with type II endometrial cancer. Int. J. Gynecol. Cancer 2019, 29 (Suppl. S4), A299. [Google Scholar]
- Zarogianni, E.; Panas, P.; Styliara, I.; Michail, G.; Koumoundourou, D.; Ravazoula, P.; Adonakis, G.; Androutsopoulos, G. EGF system receptors status in aggressive subtypes of endometrial cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 2022, 270, e86. [Google Scholar] [CrossRef]
- Morrison, C.; Zanagnolo, V.; Ramirez, N.; Cohn, D.; Kelbick, N.; Copeland, L.; Maxwell, G.; Fowler, J. HER-2 is an independent prognostic factor in endometrial cancer: Association with outcome in a large cohort of surgically staged patients. J. Clin. Oncol. 2006, 24, 2376–2385. [Google Scholar] [CrossRef]
- Engelsen, I.; Stefansson, I.; Beroukhim, R.; Sellers, W.; Meyerson, M.; Akslen, L.; Salvesen, H. HER-2/neu expression is associated with high tumor cell proliferation and aggressive phenotype in a population based patient series of endometrial carcinomas. Int. J. Oncol. 2008, 32, 307–316. [Google Scholar] [CrossRef]
- Coronado, P.; Vidart, J.; Lopez-asenjo, J.; Fasero, M.; Furio-bacete, V.; Magrina, J.; Escudero, M. P53 overexpression predicts endometrial carcinoma recurrence better than HER-2/neu overexpression. Eur. J. Obstet. Gynecol. Reprod. Biol. 2001, 98, 103–108. [Google Scholar] [CrossRef]
- Halperin, R.; Zehavi, S.; Habler, L.; Hadas, E.; Bukovsky, I.; Schneider, D. Comparative immunohistochemical study of endometrioid and serous papillary carcinoma of endometrium. Eur. J. Gynaecol. Oncol. 2001, 22, 122–126. [Google Scholar]
- Santin, A.; Bellone, S.; Siegel, E.; Palmieri, M.; Thomas, M.; Cannon, M.; Kay, H.; Roman, J.; Burnett, A.; Pecorelli, S. Racial differences in the overexpression of epidermal growth factor type II receptor (HER2/neu): A major prognostic indicator in uterine serous papillary cancer. Am. J. Obstet. Gynecol. 2005, 192, 813–818. [Google Scholar] [CrossRef]
- Santin, A.; Bellone, S.; Van Stedum, S.; Bushen, W.; Palmieri, M.; Siegel, E.; De Las Casas, L.; Roman, J.; Burnett, A.; Pecorelli, S. Amplification of c-erbB2 oncogene: A major prognostic indicator in uterine serous papillary carcinoma. Cancer 2005, 104, 1391–1397. [Google Scholar] [CrossRef]
- Slomovitz, B.; Broaddus, R.; Burke, T.; Sneige, N.; Soliman, P.; Wu, W.; Sun, C.; Munsell, M.; Gershenson, D.; Lu, K. Her-2/neu overexpression and amplification in uterine papillary serous carcinoma. J. Clin. Oncol. 2004, 22, 3126–3132. [Google Scholar] [CrossRef]
- Grushko, T.; Filiaci, V.; Mundt, A.; Ridderstrale, K.; Olopade, O.; Fleming, G. An exploratory analysis of HER-2 amplification and overexpression in advanced endometrial carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 108, 3–9. [Google Scholar] [CrossRef]
- Lukes, A.; Kohler, M.; Pieper, C.; Kerns, B.; Bentley, R.; Rodriguez, G.; Soper, J.; Clarke-Pearson, D.; Bast, R., Jr.; Berchuck, A. Multivariable analysis of DNA ploidy, p53, and HER-2/neu as prognostic factors in endometrial cancer. Cancer 1994, 73, 2380–2385. [Google Scholar] [CrossRef]
- Odicino, F.; Bignotti, E.; Rossi, E.; Pasinetti, B.; Tassi, R.; Donzelli, C.; Falchetti, M.; Fontana, P.; Grigolato, P.; Pecorelli, S. HER-2/neu overexpression and amplification in uterine serous papillary carcinoma: Comparative analysis of immunohistochemistry, real-time reverse transcription-polymerase chain reaction, and fluorescence in situ hybridization. Int. J. Gynecol. Cancer 2008, 18, 14–21. [Google Scholar] [CrossRef]
- Togami, S.; Sasajima, Y.; Oi, T.; Ishikawa, M.; Onda, T.; Ikeda, S.; Kato, T.; Tsuda, H.; Kasamatsu, T. Clinicopathological and prognostic impact of human epidermal growth factor receptor type 2 (HER2) and hormone receptor expression in uterine papillary serous carcinoma. Cancer Sci. 2012, 103, 926–932. [Google Scholar] [CrossRef]
- Díaz-Montes, T.; Ji, H.; Smith Sehdev, A.; Zahurak, M.; Kurman, R.; Armstrong, D.; Bristow, R. Clinical significance of Her-2/neu overexpression in uterine serous carcinoma. Gynecol. Oncol. 2006, 100, 139–144. [Google Scholar] [CrossRef]
- Androutsopoulos, G.; Styliara, I.; Zarogianni, E.; Michail, G.; Adonakis, G. Is it time to reconsider the clinical role of ErbB targeted therapy in endometrial cancer? In Endometrial Cancer; Farghaly, S., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2022; pp. 299–327. [Google Scholar]
- Villella, J.; Cohen, S.; Smith, D.; Hibshoosh, H.; Hershman, D. HER-2/neu overexpression in uterine papillary serous cancers and its possible therapeutic implications. Int. J. Gynecol. Cancer 2006, 16, 1897–1902. [Google Scholar] [CrossRef]
- Santin, A.; Bellone, S.; Roman, J.; McKenney, J.; Pecorelli, S. Trastuzumab treatment in patients with advanced or recurrent endometrial carcinoma overexpressing HER2/neu. Int. J. Gynaecol. Obstet. 2008, 102, 128–131. [Google Scholar] [CrossRef]
- Elsahwi, K.; Santin, A. ErbB2 overexpression in uterine serous cancer: A molecular target for trastuzumab therapy. Obstet. Gynecol. Int. 2011, 2011, 128295. [Google Scholar] [CrossRef]
- Androutsopoulos, G.; Michail, G.; Adonakis, G.; Decavalas, G. Molecular biology, expression and clinical significance of ErbB receptors in endometrial cancer. Hell. J. Obst. Gynecol. 2014, 13, 77–83. [Google Scholar]
- Fader, A.; Roque, D.; Siegel, E.; Buza, N.; Hui, P.; Abdelghany, O.; Chambers, S.; Secord, A.; Havrilesky, L.; O’Malley, D.; et al. Randomized Phase II Trial of Carboplatin-Paclitaxel Versus Carboplatin-Paclitaxel-Trastuzumab in Uterine Serous Carcinomas That Overexpress Human Epidermal Growth Factor Receptor 2/neu. J. Clin. Oncol. 2018, 36, 2044–2051. [Google Scholar] [CrossRef]
- Fader, A.; Roque, D.; Siegel, E.; Buza, N.; Hui, P.; Abdelghany, O.; Chambers, S.; Secord, A.; Havrilesky, L.; O’Malley, D.; et al. Randomized Phase II Trial of Carboplatin-Paclitaxel Compared with Carboplatin-Paclitaxel-Trastuzumab in Advanced (Stage III-IV) or Recurrent Uterine Serous Carcinomas that Overexpress Her2/Neu (NCT01367002): Updated Overall Survival Analysis. Clin. Cancer Res. 2020, 26, 3928–3935. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| ErbB-1 | ErbB-2 | ErbB-3 | ErbB-4 | |
|---|---|---|---|---|
| EGF | + | - | - | - |
| TGF-a | + | - | - | - |
| Amphiregulin | + | - | - | - |
| HB-EGF | + | - | - | + |
| Betacellulin | + | - | - | + |
| Epigen | + | - | - | + |
| Epiregulin | + | - | - | + |
| Neuregulin-1 | - | - | + | + |
| Neuregulin-2 | - | - | + | + |
| Neuregulin-3 | - | - | - | + |
| Neuregulin-4 | - | - | - | + |
| Neuroglycan C | - | - | + | - |
| Tomoregulin | - | - | - | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Androutsopoulos, G.; Styliara, I.; Zarogianni, E.; Lazurko, N.; Valasoulis, G.; Michail, G.; Adonakis, G. The ErbB Signaling Network and Its Potential Role in Endometrial Cancer. Epigenomes 2023, 7, 24. https://doi.org/10.3390/epigenomes7040024
Androutsopoulos G, Styliara I, Zarogianni E, Lazurko N, Valasoulis G, Michail G, Adonakis G. The ErbB Signaling Network and Its Potential Role in Endometrial Cancer. Epigenomes. 2023; 7(4):24. https://doi.org/10.3390/epigenomes7040024
Chicago/Turabian StyleAndroutsopoulos, Georgios, Ioanna Styliara, Evgenia Zarogianni, Nadia Lazurko, George Valasoulis, Georgios Michail, and Georgios Adonakis. 2023. "The ErbB Signaling Network and Its Potential Role in Endometrial Cancer" Epigenomes 7, no. 4: 24. https://doi.org/10.3390/epigenomes7040024
APA StyleAndroutsopoulos, G., Styliara, I., Zarogianni, E., Lazurko, N., Valasoulis, G., Michail, G., & Adonakis, G. (2023). The ErbB Signaling Network and Its Potential Role in Endometrial Cancer. Epigenomes, 7(4), 24. https://doi.org/10.3390/epigenomes7040024