
A DFT Characterization of Structural, Mechanical, and Thermodynamic Properties of Ag9In4 Binary Intermetallic Compound


Abstract
:1. Introduction
2. Computational Method and Details
3. Results and Discussion
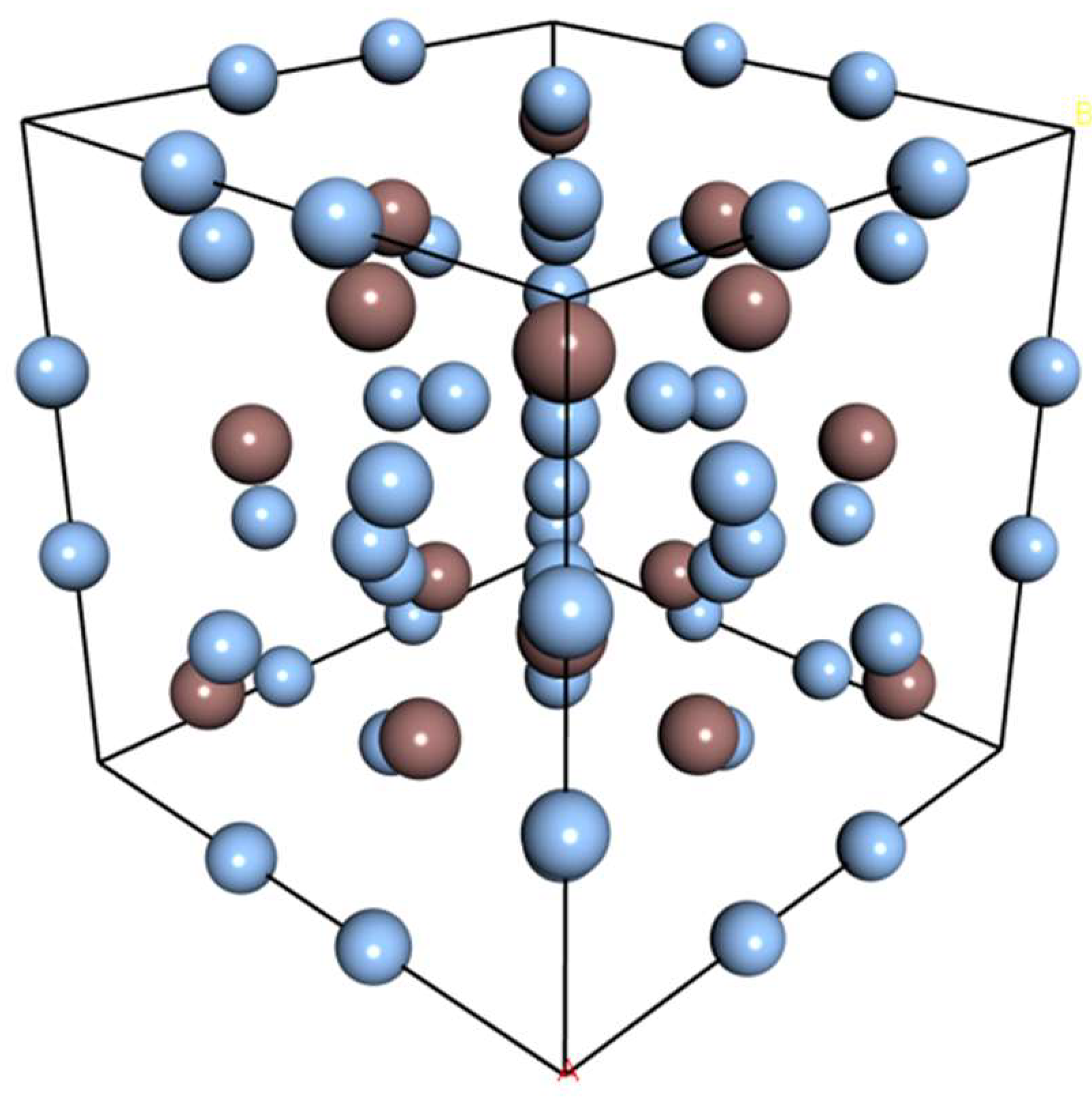
3.1. Structural Properties
3.2. Elastic and Mechanical Properties
3.3. Characterization of Elastic Anisotropic Properties
3.4. Thermodynamic Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chin, H.S.; Cheong, K.Y.; Ismail, A.B. Review on Die Attach Materials for SiC-Based High-Temperature Power Devices. Metall. Mater. Trans. B 2010, 41, 824–832. [Google Scholar] [CrossRef]
- Yuan, X.; Laird, I.; Walder, S. Opportunities, Challenges, and Potential Solutions in the Application of Fast-Switching SiC Power Devices and Converters. IEEE Trans. Power Electron. 2021, 36, 3925–3945. [Google Scholar] [CrossRef]
- Fazel, M.A.; Liyana, N.K.; Rubaiee, S.; Anas, A. A critical review on performance, microstructure and corrosion resistance of Pb-free solders. Measurement 2019, 134, 897–907. [Google Scholar] [CrossRef]
- Cheng, H.C.; Wu, Z.D.; Liu, Y.C. Viscoelastic warpage modeling of fan-out wafer level packaging during wafer-level mold cure process. IEEE Trans. Compon. Packag. Manuf. Technol. 2020, 10, 1240–1250. [Google Scholar] [CrossRef]
- Ahn, B. Emerging Interconnection Technology and Pb-Free Solder Materials for Advanced Microelectronic Packaging. Metals 2021, 11, 1941. [Google Scholar] [CrossRef]
- Deshpande, M.; Chaudhari, R.; Narayanan, P.R.; Kale, H. Evaluation of Shear Properties of Indium Solder Alloys for Cryogenic Applications. J. Mater. Eng. Perform. 2021, 30, 7958–7966. [Google Scholar] [CrossRef]
- Huang, L.C.; Zhang, Y.P.; Chen, C.M.; Huang, L.Y.; Wang, Y.P. Intermetallic compound formation and growth behavior at the interface between indium and Au/Ni(V) metallization. Mater. Charact. 2022, 184, 111673. [Google Scholar] [CrossRef]
- Huang, L.C.; Zhang, Y.P.; Chen, C.M.; Huang, L.Y.; Wang, Y.P. Interfacial reactions between pure indium solder and Au/Ni metallization. J. Mater. Sci. Mater. 2022, 33, 13143–13151. [Google Scholar] [CrossRef]
- Yang, C.A.; Kao, C.R.; Nishikawa, H.; Lee, C.C. High Reliability Sintered Silver-Indium Bonding with Anti-Oxidation Property for High Temperature Applications. In Proceedings of the Electronic Components and Technology Conference, San Diego, CA, USA, 28 May–1 June 2018. [Google Scholar]
- Cheng, H.-C.; Tai, L.-C.; Liu, Y.-C. Theoretical and Experimental Investigation of Warpage Evolution of Flip Chip Package on Packaging during Fabrication. Materials 2021, 14, 4816. [Google Scholar] [CrossRef]
- Wang, Y.P.; Liu, H.B.; Bao, M.D.; Zhou, B.; Wu, H.C. Interfacial Reactions of Sn-3.0Ag-0.5Cu Solder with Sputter Cu-Ti Alloy Film UBM. J. Electron. Mater. 2021, 50, 3692–3698. [Google Scholar] [CrossRef]
- Chang, J.S.; Chen, S.W. Liquidus Projection and Isothermal Section of the Ag-In-Zn Ternary System. J. Electron. Mater. 2015, 44, 1134–1143. [Google Scholar] [CrossRef]
- Song, J.M.; Lu, W.C.; Chou, P.W. Nanomechanical Responses of an Intermetallic Compound Layer in Transient Liquid Phase Bonding Using Indium. J. Electron. Mater. 2020, 49, 18–25. [Google Scholar] [CrossRef]
- Wang, P.J.; Sha, C.H.; Lee, C.C. Silver Microstructure Control for Fluxless Bonding Success Using Ag-In System. IEEE Trans. Compon. Packag. Manuf. Technol. 2010, 33, 462–469. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Nwoke, D.; Barlow, F.D.; Lee, C.C. Thermal Cycling Reliability Study of Ag–In Joints Between Si Chips and Cu Substrates Made by Flux less Processes. IEEE Trans. Compon. Packag. Manuf. Technol. 2014, 4, 1420–1426. [Google Scholar]
- Qin, H.B.; Zhang, X.P.; Zhou, M.B.; Li, X.P.; Mai, Y.W. Geometry effect on mechanical performance and fracture behavior of micro-scale ball grid array structure Cu/Sn–3.0Ag–0.5Cu/Cu solder joints. Microelectron. Reliab. 2015, 55, 1214–1225. [Google Scholar] [CrossRef]
- Cheng, H.C.; Cheng, H.K.; Lu, S.T.; Chen, W.H. Drop Impact Reliability Analysis of 3-D Chip-on-Chip Packaging: Numerical Modeling and Experimental Validation. IEEE Trans. Device Mater. Reliab. 2014, 14, 499–511. [Google Scholar] [CrossRef]
- Hsu, S.J.; Lee, C.C. Bonding of SiC Chips to Copper Substrates Using Ag-In System. In Proceedings of the Electronic Components and Technology Conference, San Diego, CA, USA, 26–29 May 2015. [Google Scholar]
- Lin, W.P.; Lee, C.C. Fluxless Bonding of Bismuth Telluride Chips to Alumina Using Ag–In System for High Temperature Thermoelectric Devices. IEEE Trans. Compon. Packag. Manuf. Technol. 2011, 1, 1311–1318. [Google Scholar] [CrossRef]
- Inukai, M.; Soda, K.; Sato, H.; Mizutani, U. Electronic structure of Ag5Zn8, Ag9In4 and Mn3In gamma-brasses studied by FLAPW band calculations. Philos. Mag. 2011, 91, 2543–2547. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Shih, Y.C.; Chen, C.S.; Wu, K.C. Chain Length Effect on Surface Stress of Alkanethiolates Adsorbed onto Au (111) Surface: A van der Waals Density Functional Study. J. Mech. 2014, 30, 241–246. [Google Scholar] [CrossRef]
- Brandon, J.K.; Brizard, R.Y.; Pearson, W.B.; Tozer, D.J.N. γ-Brasses with I and P cells. Acta Cryst. 1977, B33, 527–537. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Wang, W.; Yang, C.; Bai, L.; Li, M.; Li, W. First-Principles Study on the Structural and Electronic Properties of Monolayer MoS2 with S-Vacancy under Uniaxial Tensile Strain. Nanomaterials 2018, 8, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, B.; Shahrokhi, M.; Madjet, M.; Hussain, T.; Zhuang, X.; Rabczuk, T. N-, B-, P-, Al-, As-, and Ga-graphdiyne/graphyne lattices: First-principles investigation of mechanical, optical and electronic properties. J. Mater. Chem. C 2019, 7, 3025–3036. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y. The structural stability and optical properties of NiPt nanomaterial from first-principles investigations. Mater. Sci. Semicond. Process. 2020, 120, 105306. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, Y.; Zhao, Y.; Ma, Y. Anisotropies in Elasticity, Sound Velocity, and Minimum Thermal Conductivity of Low Borides VxBy Compounds. Metals 2021, 11, 577. [Google Scholar] [CrossRef]
- Lin, Y.; Chong, X.; Ding, Y.; Zhou, Y.; Gan, M.; Xu, L.; Wei, S.; Feng, J. First-Principles Calculations of Thermal and Electrical Transport Properties of bcc and fcc Dilute Fe–X (X = Al, Co, Cr, Mn, Mo, Nb, Ni, Ti, V and W) Binary Alloys. Metals 2021, 11, 1988. [Google Scholar] [CrossRef]
- Fang, C.; Souissi, M.; Que, Z.; Fan, Z. Crystal Chemistry and Electronic Properties of the Al-Rich Compounds, Al2Cu, ω-Al7Cu2Fe and θ-Al13Fe4 with Cu Solution. Metals 2022, 12, 329. [Google Scholar] [CrossRef]
- Wang, J.; Qin, H.; Chen, J.; Yang, D.; Zhang, G. First-Principles Study on the Elastic Mechanical Properties and Anisotropies of Gold–Copper Intermetallic Compounds. Metals 2022, 12, 959. [Google Scholar] [CrossRef]
- Li, B.; Duan, Y.; Peng, M.; Shen, L.; Qi, H. Anisotropic Elastic and Thermal Properties of M2InX (M = Ti, Zr and X = C, N) Phases: A First-Principles Calculation. Metals 2022, 12, 1111. [Google Scholar] [CrossRef]
- Tang, Q.; Yue, Y.; Jiang, Z.; Li, F.; Wang, T.; Jin, C.; Dong, H.; Chang, R.; Wang, B.; Cai, H.; et al. Non-metallic carbon-based catalysts for acetylene hydrochlorination: The effect of graphitization degree of carbonaceous material. Catal. Commun. 2022, 167, 106458. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Head, J.D.; Zerner, M.C. A Broyden—Fletcher—Goldfarb—Shanno optimization procedure for molecular geometries. Chem. Phys. Lett. 1985, 122, 264–270. [Google Scholar] [CrossRef]
- Brillouin, L. Les électrons dans les métaux et le classement des ondes de de Broglie correspondantes. Comptes Rendus Hebd. Des Séances De L′Académie Des Sci. 1930, 191, 292–294. [Google Scholar]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Nye, J.F. Physical Properties of Crystals; Oxford University Press: Oxford, UK, 1985. [Google Scholar]
- Marmier, A.; Lethbridge, Z.A.D.; Walton, R.I.; Smith, C.W.; Parker, S.C.; Evans, K.E. ElAM: A computer program for the analysis and representation of anisotropic elastic properties. Comput. Phys. Commun. 2010, 181, 2102–2115. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, N.W.; Mermin, N.D. Solid State Physics; Saunders College: Philadelphia, PA, USA, 1976. [Google Scholar]
- Pettifor, D.G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 1992, 8, 345–349. [Google Scholar] [CrossRef]
- Jund, P.; Viennois, R.; Tao, X.; Niedziolka, K.; Tédenac, J.C. Physical properties of thermoelectric zinc antimonide using first-principles calculations. Phys. Rev. B 2012, 85, 224105. [Google Scholar] [CrossRef] [Green Version]
- Verma, J.K.D.; Nag, B.D. On the Elastic Moduli of a Crystal and Voigt and Reuss Relations. J. Phys. Soc. Jpn. 1965, 20, 635–636. [Google Scholar] [CrossRef]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Cheng, H.C.; Cheng, T.-H.; Chen, W.-H.; Chang, T.-C.; Huang, H.-Y. Board-Level Drop Impact Reliability of Silicon Interposer-Based 2.5-D IC Integration. IEEE Trans. Compon. Packag. Manuf. Technol. 2016, 6, 1493–1504. [Google Scholar] [CrossRef]
- Teter, D.M. Computational Alchemy: The Search for New Superhard Materials. MRS Bull. 1998, 23, 22–27. [Google Scholar] [CrossRef]
- Chen, X.Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.J.; Xu, B.; Zhao, Z.S. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Met. Hard Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Liu, Z.T.Y.; Gall, D.; Khare, S.V. Electronic and bonding analysis of hardness in pyrite-type transition-metal pernitrides. Phys. Rev. B 2014, 90, 134102. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [Green Version]
- Vahldiek, F.W.; Mersol, S.A. Anisotropy in Single-Crystal Refractory Compounds; Springer: New York, NY, USA, 1968. [Google Scholar]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Debye, P. Zur Theorie der spezifischen Wärmen. Ann. Phys. Leipz 1912, 344, 789–839. [Google Scholar] [CrossRef] [Green Version]
- Petit, A.T.; Dulong, P.L. Recherches sur quelques points importants de la Théorie de la Chaleur. Ann. Chim. Phys. 1819, 10, 395–413. [Google Scholar]
- Cahill, D.G.; Watson, S.K.; Pohl, R.O. Lower limit to the thermal conductivity of disordered crystals. Phys. Rev. B 1992, 46, 6131–6140. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | x | y | z |
|---|---|---|---|
| Ag | 0.605 | 0.605 | 0.605 |
| Ag | −0.162 | −0.162 | −0.162 |
| Ag | 0.318 | 0.318 | 0.318 |
| Ag | 0 | 0 | 0.355 |
| Ag | 0.5 | 0.5 | 0.854 |
| Ag | 0.320 | 0.320 | 0.034 |
| In | 0.122 | 0.122 | 0.122 |
| In | 0.808 | 0.808 | 0.530 |
| Atom | x | y | z |
|---|---|---|---|
| Ag | 0.6049 | 0.6049 | 0.6049 |
| Ag | −0.1699 | −0.1699 | −0.1699 |
| Ag | 0.3259 | 0.3259 | 0.3259 |
| Ag | 0 | 0 | 0.3563 |
| Ag | 0.5 | 0.5 | 0.8556 |
| Ag | 0.3179 | 0.3179 | 0.02834 |
| In | 0.1236 | 0.1236 | 0.1236 |
| In | 0.8103 | 0.8103 | 0.5331 |
| Mechanical Properties | Lattice Constants (Å) | V(Å3) | C11 | C44 | C12 | PCauchy | K | G | E | Hv | K/G | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Present study | a = b = c = 10.029 | 1008.73 | 128.6 | 28.2 | 54.9 | 26.7 | 79.47 | 31.37 | 83.17 | 3.67 | 0.3 | 2.53 |
| Experiment [24] | a = b = c = 10.037 | 1011.14 | - | - | - | - | - | - | - | - | - | - |
| Experiment [13] | - | - | - | - | - | - | - | - | 89.4 ± 2.2 | 3.2 ± 0.22 | - | - |
| Cu6Sn5 [13] | - | - | - | - | - | - | - | - | 110 ± 3.1 | 7.3 ± 0.08 | - | - |
| Cu3Sn [13] | - | - | - | - | - | - | - | - | 140 ± 2.1 | 7.2 ± 0.21 | - | - |
| Cu5Zn8 [13] | - | - | - | - | - | - | - | - | 170 ± 2.2 | 6.9 ± 0.09 | - | - |
| Ni3Sn4 [13] | - | - | - | - | - | - | - | - | 127 ± 2.9 | 4.5 ± 0.13 | - | - |
| AuZn3 [13] | - | - | - | - | - | - | - | - | 111 ± 2.3 | 2.2 ± 0.07 | - | - |
| Cu11In9 [13] | - | - | - | - | - | - | - | - | 109.5 ± 0.84 | 6.57 ± 0.15 | - | - |
| Anisotropic Index | AU | AB (%) | AG (%) |
|---|---|---|---|
| Value | 0.088 | 0 | 0.87 |
| Young’s Modulus | Whole | yz | xz | xy |
|---|---|---|---|---|
| Emax (GPa) | 95.78 | 95.78 | 95.78 | 95.78 |
| Emin (GPa) | 75.56 | 79.78 | 79.78 | 79.78 |
| Shear Modulus | Whole | yz | xz | xy |
|---|---|---|---|---|
| Gmax (GPa) | 36.86 | 28.16 | 28.16 | 28.16 |
| Gmin (GPa) | 28.16 | 28.16 | 28.16 | 28.16 |
| Property | ρ (Kg/m3) | (m/s) | (m/s) | kmin (W/(m·K)) |
|---|---|---|---|---|
| Value | 9389.8 | 3594.2 | 1827.8 | 0.544 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, H.-C.; Yu, C.-F. A DFT Characterization of Structural, Mechanical, and Thermodynamic Properties of Ag9In4 Binary Intermetallic Compound. Metals 2022, 12, 1852. https://doi.org/10.3390/met12111852
Cheng H-C, Yu C-F. A DFT Characterization of Structural, Mechanical, and Thermodynamic Properties of Ag9In4 Binary Intermetallic Compound. Metals. 2022; 12(11):1852. https://doi.org/10.3390/met12111852
Chicago/Turabian StyleCheng, Hsien-Chie, and Ching-Feng Yu. 2022. "A DFT Characterization of Structural, Mechanical, and Thermodynamic Properties of Ag9In4 Binary Intermetallic Compound" Metals 12, no. 11: 1852. https://doi.org/10.3390/met12111852
APA StyleCheng, H.-C., & Yu, C.-F. (2022). A DFT Characterization of Structural, Mechanical, and Thermodynamic Properties of Ag9In4 Binary Intermetallic Compound. Metals, 12(11), 1852. https://doi.org/10.3390/met12111852