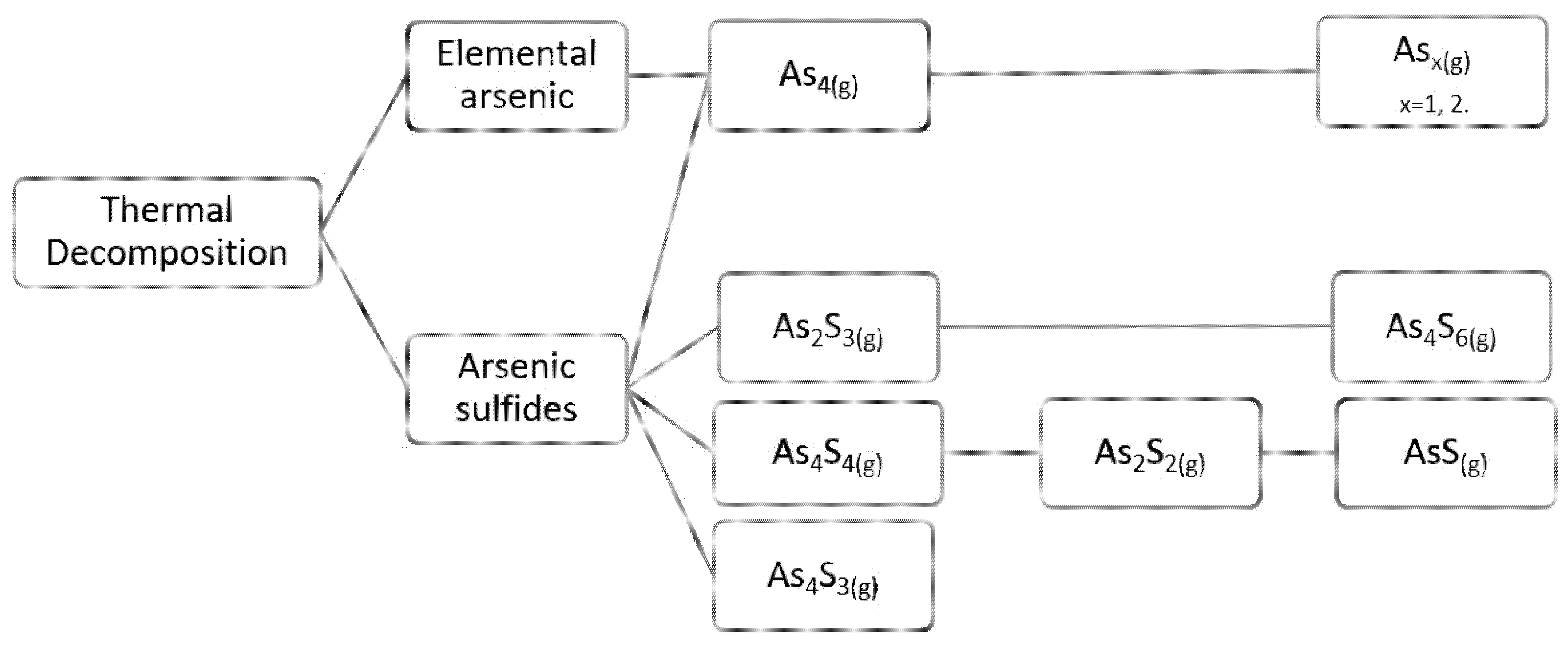
3.1. Elemental Arsenic
Studies related to the reaction mechanisms during thermal decomposition and oxidation of elemental arsenic are very scarce. However, the existing information is related to the volatilization and oxidation of arsenic from arsenical minerals or copper concentrates treated under pyrometallurgical processes.
In 1979, Weisenberg and Bakshi [
22] analyzed arsenic removal in copper smelting for different types of reaction furnaces, intending to organize the information present to date on the distribution and control of arsenic in the main copper smelting systems in the United States; there was no experimental work. The authors emphasized the scarcity and inconsistency of the information present on arsenic distribution in the literature analyzed, provided by The Environmental Protection Agency; of the information that was studied by the authors they emphasize the amount of arsenic in the feed, since in most U.S. smelters it was generally below 1.0 wt.%, and this value was not constant; therefore, due to the small amount of arsenic present in the feed, in many cases, it cannot be adequately measured. In addition, they noted that changes in smelting techniques and equipment tend to produce variations in arsenic distribution; and, that the presence of other elements such as oxygen or copper within the smelting process can directly influence arsenic distribution. According to the authors, arsenic in copper smelting was removed by volatilization in elemental or sulfide form (specific phases are not reported), and subsequently oxidized in the presence of oxygen to gaseous As
2O
3 which upon cooling condenses and is collected as residual dust. However, in an oxidizing atmosphere, the trioxide formed oxidizes to As
2O
5, which is less volatile and forms non-volatile arsenates of calcium or iron.
For the distribution and control of arsenic in different reactors, the authors mention that the sulfides present in the copper concentrate that need to be oxidized by fluidized bed roasting are the FeS, As
2S
3, As
2S
3, and As
2S
5 phases; and the arsenic removal was by its volatilization which depends on the temperature, residence time and the type of atmosphere. According to the authors, in order to explain the temperature used during roasting, they mention that As
2S
3 has its boiling point at 707 °C, whereas As
2S
5 sublimates at 500 °C through decomposition; meanwhile, As
2O
3 melts at 310 °C and boils at 457 °C. Consequently, if copper concentrates are heated above 700 °C, arsenic removal should be assured. However, the copper concentrate has a large amount of sulfur, and high temperatures, favorable for arsenic removal, are not consistent for sulfur removal, since at the beginning of roasting a large part of easily meltable sulfides are present, a high temperature would melt the particles, which would reduce the surface-to-volume ratio for sulfur removal; however, if a low temperature—not specified by the author—is maintained during the whole roasting process, sulfates would be formed, because the sulfates’ formation heat would be higher than that of their oxides; for example, for the case of FeSO
4 and Fe
3O
4 formation from FeS at 400 °C, heat of formation value is −835.25 kJ and −1731.87 kJ, respectively [
47]. Therefore, the ideal condition proposed by the authors was to start with low temperatures and gradually increase up to the roasting temperature. Finally, to conclude their discussion on roasting, the authors propose that arsenic removal by reductive roasting would be feasible because the maximum As removal takes place between 498 and 593 °C under reductive conditions; however, oxidative roasting is required to remove sulfur. Therefore, alternating between oxidizing and reducing processes several times during roasting could maximize the volatilization of arsenic and sulfur, which can be developed in a multi-deck roaster as opposed to a fluidized bed that allows only one of these conditions.
At the date of publication, 1979, most U.S. smelters were using the reverberatory furnace for copper smelting, but the trend in newer or modified smelters is to smelt instantaneously, in molten bath (Noranda, SKS, Ausmelt, Teniente Converter, others) or electric furnace smelting. This change results in reduced production costs and increased SO2 concentration for environmental control; in the reverberatory furnace, arsenic is mainly removed by volatilization and as a slag constituent. If the arsenic fed is greater than 0.2%, it volatilizes between 55 and 75%, and 10–25% of it is slagged, whereas if arsenic is present in smaller quantities, it volatilizes between 5 and 37% and is slagged between 16–55%.
Regarding an electric furnace, the authors mention that the behavior of arsenic is similar to that of a reverberatory furnace, whereas in a flash furnace, Outokumpu type, the amount of volatilized arsenic varies between 76 and 85%, and the sorbed arsenic varies between 7 and 17%.
The authors emphasize that the behavior of arsenic in the melting furnaces is controlled by the fact that the reactions occur in the presence of copper, since As is more stable in Cu than in Cu2S, due to a decrease in its thermodynamic activity and, therefore, in its volatilization; therefore, the behavior of arsenic in melting processes varies with the amount of matte produced.
Because of the above, in the Noranda converter, arsenic removal is favorable at low matte grades. Since arsenic volatilization plays an important role in its removal during matte production in melting furnaces, any factor that increases volatilization will further enhance arsenic removal. For example, mineralization, some As compounds have higher vapor pressures than others and will therefore volatilize to a greater degree as well as the temperature of the reactor/melting furnace; and the exposure of the slag surface, the greater the exposure of the slag to the atmosphere, the greater the volatilization of As from the slag. The molten matte contains Cu, Fe, and S as its main components with up to 3% dissolved oxygen. In addition, it contains minor amounts of impurities such as As, Bi, Pb, etc., which are not removed during smelting. The distribution of As in a copper converter between gases and slag varies widely. When converting a high-grade matte, metallic copper appears early in the cycle and acts as an As collector because As, as mentioned above, is more stable and less volatile in metallic copper than in copper sulfides. Arsenic entering the copper smelting process is volatilized and removed as metal oxides in the gas streams, or with the slag. The metal oxides reported in the gases are the result of very high temperatures associated with the pyrometallurgical processes used and the inherent volatility of As and As2O3.
Finally, they conclude that the arsenic distribution is controlled by the properties of the arsenic and its environment, and that, by using two types of concentrates, characterized by their arsenic content by weight, different distributions are generated, those of high input level (As > 0.2%), where the distribution of arsenic is highly influenced by temperature and the presence of copper; and those of low level (As < 0.2%) where arsenic is not so easily released and passes to the slag.
Nakazawa, Yazawa, and Jorgensen in 1999 [
30] studied the removal of sulfide arsenic from copper concentrate, this work was studied in detail in the section on As-S in neutral atmosphere. From it, is clear that, for high oxidation degrees, volatilizations of elemental arsenic and its sulfides are gradually replaced by As
4O
6. In addition, the total removal of arsenic during roasting depends directly on the temperature and the degree of oxidation used; the lower the amount of arsenic present in the system, the more difficult it is to remove it using the roasting process.
According to Weisenberg et al. [
22], the volatilizations of arsenic under an oxidizing atmosphere are in the elemental or sulfurized form; and, subsequently, these are oxidized in the gas phase to As
2O
3(g); while Nakazawa et al. [
30] likewise indicate that the volatilizations are in the elemental or sulfurized form; however, they present the As
4O
6(g) phase as the predominant final phase when the oxygen concentration increases. Therefore, it can be concluded that the As
2O
3(g), generated during the oxidation of the volatilized gas phase, forms the As
4O
6(g) dimer, which is its stable form, as can be seen in
Figure 3.
3.2. Arsenic Sulfides
As mentioned above, Nakazawa et al. (1999) [
30] performed a simulation using HSC [
47] for the removal of arsenic, bismuth, and antimony during the roasting of copper concentrates, obtaining that as oxidation degree increases, elemental arsenic volatilizations and its sulfides are gradually replaced by As
4O
6. For the oxidation of As
2S
3 under 700 °C and PSO
2 = 0.1 bar, arsenic is oxidized to AsS in low oxygen atmospheres; in turn, with increasing oxygen content AsS is oxidized to the As
2O
3 form. However, at temperatures above 700 °C AsS decomposes to As and S, which subsequently oxidizes to As
2O
3. At pressures of 10–5.35 bar, non-volatile As
2O
4 is formed. When Fe
2O
3 is present in the system, arsenates are formed, which are a stable, non-volatile product, according to the following reaction:
The vapor pressure of the volatile species As, Sb, and Bi were calculated using the HSC Chemistry program [
47]; arsenic species have high vapor pressures compared with Bi and Sb volatilizations, and almost complete volatilization is expected to occur at roasting temperatures.
It is clear that Sb and Bi compounds are much less volatile than As; at low temperatures, As4O6(g) has the highest vapor pressure, however, at high temperatures As4(g) is the predominant phase; and, As2(g) predominates at melting temperatures. Due to the relatively low vapor pressure of As2S3(g), volatilization will proceed at a faster rate from AsS(s) than from As2S3(s). Species such as FeAs2O4, As2O4, and As2O5 are non-volatile.
The authors emphasize that it is well known that the removal of As from copper concentrates depends on the degree of oxidation and that the optimum conditions for its removal have not yet been established. As an example, a copper concentrate with 5.3% As, which was oxidized in air at 700 °C, was simulated in the HSC Chemistry program [
47]; obtaining that the As behavior was as follows; when the degree of oxidation is low, the predominant volatilized species were As
2S
3, As
4S
4, As
4, and As
2; which were gradually replaced by As
4O
6 as the degree of oxidation increased. When the oxygen content was set between 2400 and 3600 moles, the predominant species was As
4O
6. If there is enough oxygen in the system for all the Cu
2S in the concentrate to be oxidized, the oxygen equilibrium pressure will be high and non-volatile Fe-AsO
4 will be formed as a stable species instead of As
4O
6(g). When the amount of oxygen moles exceeded 3600, the calculations showed that all arsenic can be removed. In order to study the temperature, a 2000 moles oxygen loading was simulated between 500 and 900 °C, where it was obtained that the predominant species at low temperatures was As
4O
6 and then it is gradually replaced by As
4 and As
2.
Wasson et al. in 2005 [
27] studied the emissions of chromium, copper, and arsenic from the open burning of Chromated Copper Arsenate (CCA) treated wood. They experimentally simulated the combustion of wood waste in order to characterize the emission composition, particle size distribution of the ash, and the resulting partitioning of arsenic, copper, and chromium between the fly ash and the residual ash. CCA was one of the main wood preservatives in the United States from 1940 until 2003, when it was discontinued for this type of application; this solution was composed of CrO
3 (~47%), CuO (~19%), and As
2O
5 (~34%), depending on the wood using a retention between 4.0 and 40.0 kg/m
3 of AAC—i.e., between 4.0 and 40.0 kg of preservative absorbed per cubic meter of wood; but in general most woods were treated with retention of 6.4 kg/m
3. This type of wood is not defined as hazardous waste, so it is often burned for disposal, although this is not legally permitted. Between 20,000 and 70,000 ppm of arsenic have been found in campfire residues; in addition, approximately 20% of emissions from CCA-treated wood correspond to arsenic volatilization, approximately 1% to chromium, and copper; and at temperatures above 800 °C, arsenic volatilization of over 80% has been found.
For the experimental development of the tests, 7 kg of wood loaded with 6.4 kg/m
3 of CCA was used, which was burned for 30 min at temperatures up to 1200 °C in a box of 3 m × 2.8 m × 2.1 m with an exit duct, which had a diameter of 20.3 cm and an air extractor, where samples were taken in bags for subsequent analysis by SEM, XRD, and ICP-MS. The wood was placed in a container filled with sand on an electronic balance. Arsenic, chromium, and copper corresponded to 0.1 to 0.3% of the treated mass per sample. The resulting ash consisted of 1.3% arsenic and chromium, and 60% copper. The particle size of the resulting ash was in the range of 0.1 to 1.0 µm. The volatilization of arsenic can be affected by the presence of Fe and Ca in the wood as it forms non-volatile compounds. In this case, Fe and Ca are present in amounts between 500 and 600 ppm. They refer to simulations developed using Chemsage software [
51], which predict that large amounts of arsenic would volatilize below 600 °C, and Cu and Cr volatilizations are negligible below 1200 and 1500 °C, respectively. At 1200 °C the volatilized As phases include As
2O
5 and As
4O
6. They emphasize that the equilibrium predictions may be erroneous since the thermochemical information of the species may have low accuracy in the database, however, the information delivered by this software is of great help in explaining results and predicting experimental behavior. They obtain as a result of the emissions that the amount of As, Cr, and Cu released is in the range of 190–240, 8–22, 9–13 mg/kg CCA treated wood. The residual ash is composed of approximately 8.4% As, 15.9% Cr, and 9.2% Cu, where 14% of the arsenic contained was volatilized; part of the residual arsenic may be present as arsenates with chromium. Finally, they conclude that arsenic is present in the emissions as a mixture of As
2O
5 and As
4O
6, and the total As
3+/As ratio is estimated to vary between 0.8 and 0.9; copper is present as Cu
+ and Cu
2+, with a total Cu
+/Cu ratio between 0.65 and 0.7; finally, the chromium present in the volatilizations is as Cr
3+.
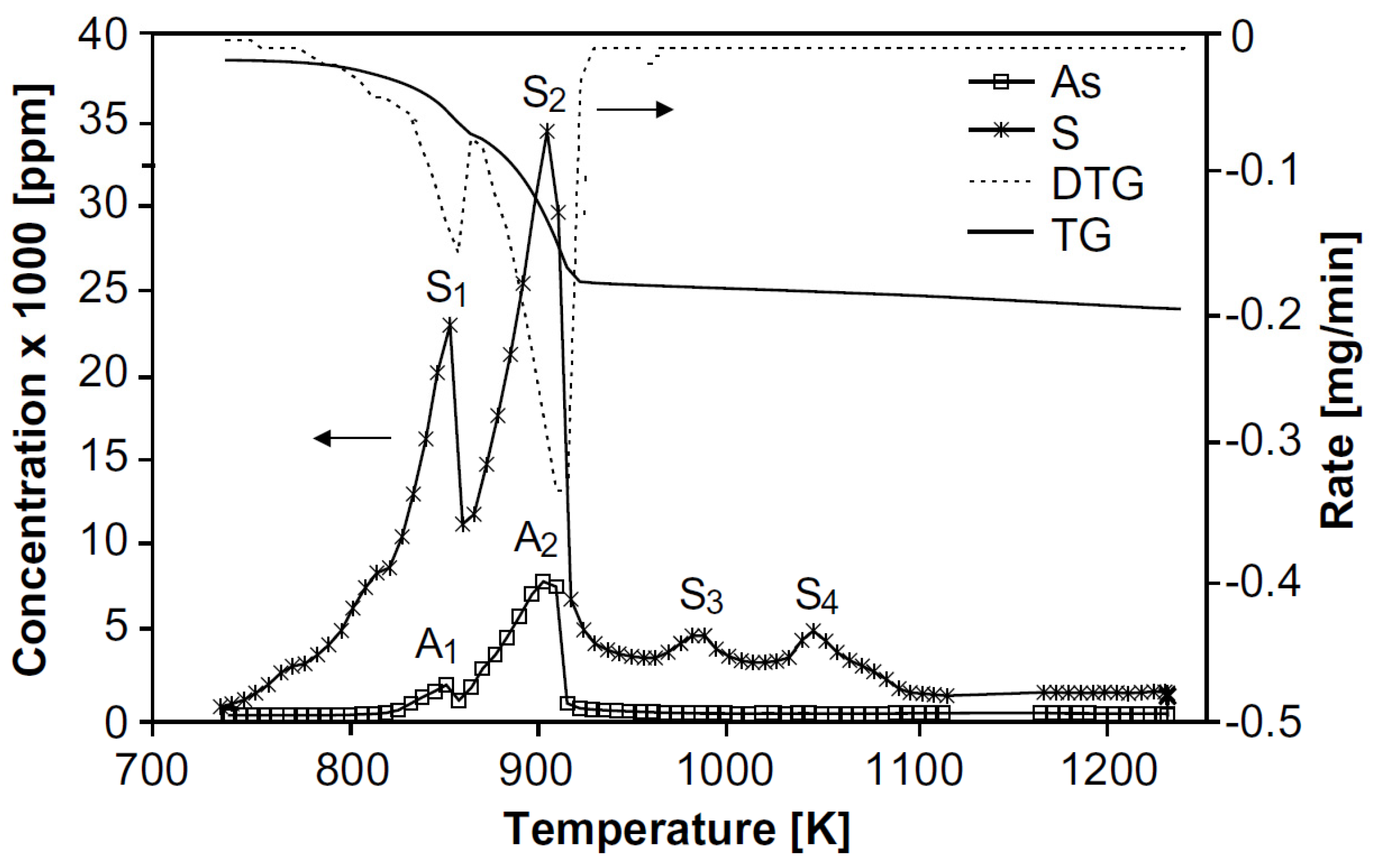
In 2005, Mihajlović et al. [
52] studied the isothermal and non-isothermal oxidation kinetics of As
2S
2, with the aim of determining the kinetic parameters and oxidation mechanisms. For this purpose, they performed experimental tests of thermal analysis, they used a synthesized sample of As
2S
2 from Paisy Hilendarsky Plovdiv University (Bulgaria) with the following composition, 67.22% As and 32.78% S; the phases were identified by XRD. Multiple DTA-TG-DTG tests were performed to investigate the oxidation process in a non-isothermal manner. These tests were carried out on the Derivatograph 1500 thermal analysis equipment, operated under atmospheric air, with a heating rate of 20 °C/min and a maximum temperature of 1000 K. While the isothermal tests were developed using standard equipment used for this purpose, where the sample was heated in a Mars oven, in which a defined airflow was injected into the reaction zone, the exhaust gases were taken to adsorption tanks filled with hydrogen peroxide where sulfuric acid was formed. The results of the DTA-TG-DTG tests are summarized in
Figure 4; the DTA curve shows the first peak in the range of 150–320 °C and a second peak between 320 and 670 °C, which are accompanied by a mass loss which is not significant until 360 °C, after which it increases significantly and substantially; this can be seen in the TG curve.
According to the information provided by the literature and the figure above, it is possible to determine that the oxidation of As
2S
2 when heated under atmospheric air proceeds as follows:
where above 193 °C, As
2O
3 forms the dimer As
4O
6(g) according to:
In parallel to the above reaction, up to 315 °C, the formation of As
2O
5 is possible according to:
Additionally, above 321 °C, oxidation occurs according to:
As mentioned, the weight loss is negligible up to 360 °C, this is because the molar mass of As
2S
2 is similar to that of As
2O
3 and As
2O
5. As the temperature increases, the solubility of sulfur decreases, and according to the information in the literature at 445 °C an equilibrium in As
4S
3 and gaseous SO
2 is possible:
This is in agreement with the results obtained by the authors by ICP for a sample heated to 350 °C, which showed a composition of As
4S
3. Furthermore, this molten phase also reacts with oxygen forming As
4O
6 and SO
2, and above 534 °C the As
4S
3 begins to volatilize and oxidize, and above 650 °C only the following reaction is possible:
According to
Figure 4, the first peak between 150 and 320 °C corresponds to the formation of As
2O
3, As
4O
6(g), and As
2O
5.
Above 360 °C, a rapid weight loss begins, which is due to the fact that As2O5(s) is not stable at high temperatures, dissociating to As4O6(g), and O2(g).
An almost linear mass loss is recorded above 534 °C up to 650 °C, which corresponds to the volatilization of As4S3.
In the second interval of the DTA curve in
Figure 4, between 320 to 670 °C an exothermic peak was reported, corresponding to the formation reactions of As
4O
6(g) from As
2S
2(l), and As
4S
3(l/g), together with the formation of SO
2(g).
The slight mass increase after 620 °C can only be explained by the formation of gaseous products that must diffuse through the remaining As4S3 liquid layer into the gas and are partially trapped as bubbles. After the entire liquid phase is evaporated the mass loss in the TG curve is completed.
Therefore, it is concluded that the oxidation of As2S2 can be represented by two stages; the first is the melting of As2S2 and its transformation to As4S3, for its subsequent oxidation to As2O5 and As4O6(g), and the second stage would correspond to the oxidation of As4S3 to As4O6(g).
For the calculation of the kinetic parameters, they used the Borchardt and Daniels’ method [
53] for a non-isothermal model, and determined an activation energy of 95 kJ/mol for the temperature range between 320 and 670 °C; while for the temperature range between 350 and 450 °C, which corresponds to the transformation of As
2S
2 into As
4S
3, the activation energy was calculated isothermally obtaining 75 kJ/mol.
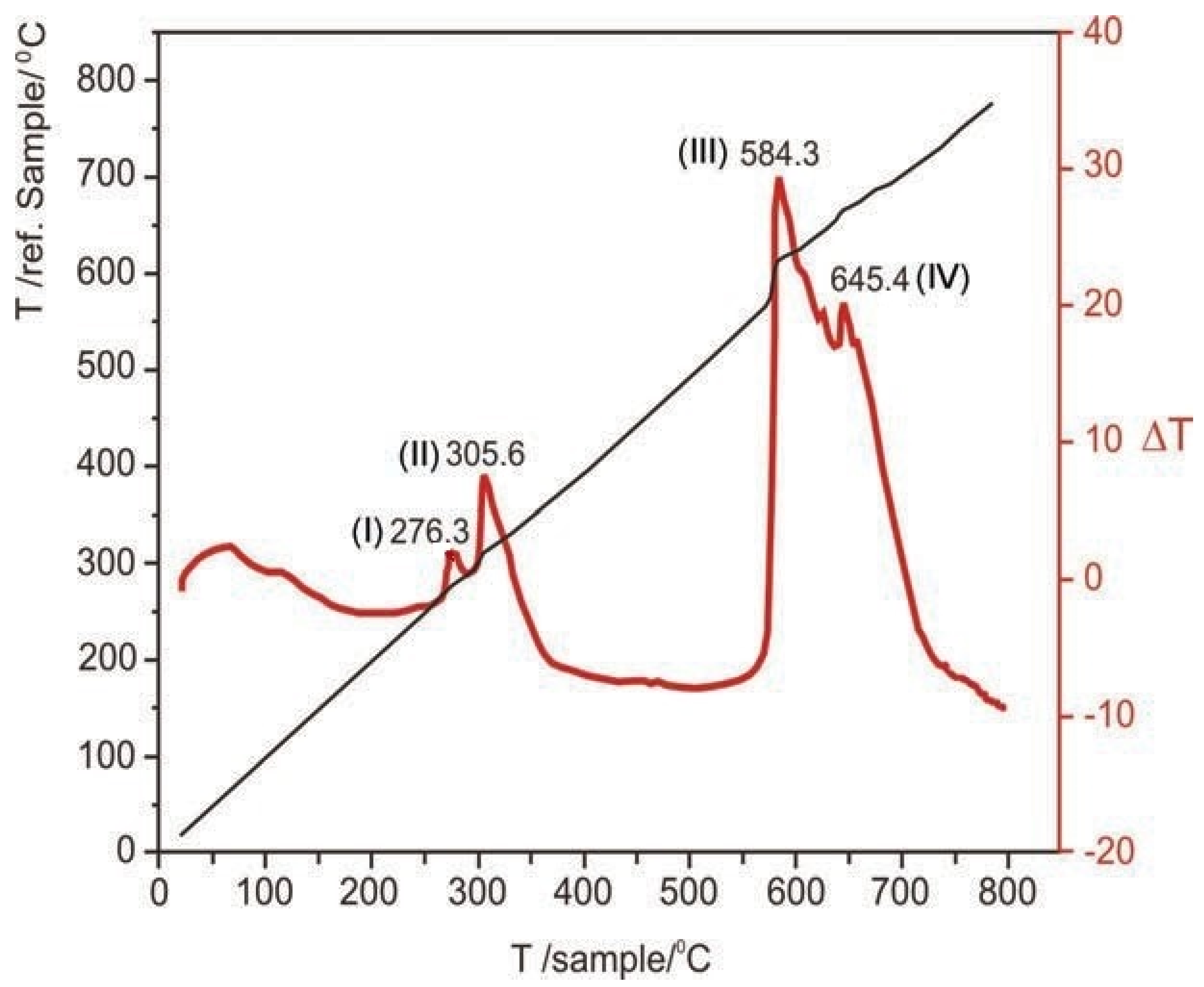
Later in 2009, Štrbac et al. [
28] experimentally studied non-isothermally a natural mixture of As
2S
3 and As
2S
2, in order to determine the kinetics and oxidation mechanism. Initially, they emphasize that the As-S system is a subject of great interest due to the insufficient information on the oxidation mechanism reactions occurring in this system, as well as the lack of complete databases with thermodynamic and kinetic parameters describing the compounds’ oxidation processes of this system. In addition, they point out that the information differs depending on the source from which they are obtained. The natural sample came from the Trepča mine, Serbia, which was characterized by XRD, obtaining that the main phases were As
2S
3, As
2S
2 together with quartz and pyrite. Non-isothermal tests were developed on Derivatograph 1500 thermal analysis equipment, with a heating rate of 20 °C/min and a maximum temperature of 1273 K. XRD analysis was performed on samples treated at 700 °C. As
2O
3 and Fe
2O
3 were detected. The DTA result obtained during the non-isothermal tests is presented in
Figure 5, where two exothermic peaks connected at 276.3 and 305.6 °C, and two other peaks connected at 584.3 and 645.4 °C can be observed. These conjoined peaks represent parallel reactions, the first set corresponds to the oxidation of the two arsenic sulfides to As
2O
3, which is volatilized mostly as As
4O
6(g); while the third peak corresponds to the volatilization and oxidation of the remaining arsenic sulfides to As
4O
6(g). The fourth peak, meanwhile, represents the oxidation of pyrite to Fe
2O
3.
For the activation energy calculation, they use the Borchardt and Daniels’ model [
53] for non-isothermal systems, obtaining for the first set of reactions comprised between 260 and 330 °C, the reaction energy of 101 and 110 kJ/mol, respectively; while for the second set comprised between 580 and 745 °C, 77 and 68 kJ/mol, respectively.
From the review developed, we can corroborate what is shown in
Figure 1, and what was mentioned above for the volatilized phases from elemental arsenic; the predominant volatilization is As
4O
6(g); however, when the degree of oxidation is low, the species As
2S
3, As
4S
4, As
4, and As
2 are present, and they oxidize to As
4O
6(g) as we increase the degree of oxidation. As
4O
6(g) is stable at low temperatures and above 900 °C is gradually replaced by As
4 and As
2(g). In addition, the possible presence of As
2O
5 in the gas phase is reported, but it is not stable and decomposes from its solid state into As
2O
3(g) and O
2(g), for the subsequent formation of the As
4O
6(g) dimer.
3.3. Arsenic Sulfide Compounds
In 2001, Welhan [
54] experimentally studied the mechanochemical processing of enargite to analyze the effect of grinding under different atmospheres on the solubility of enargite concentrate. Enargite is a copper ore containing arsenic. In smelting processes, ores with a high arsenic content are a problem due to the cost of removing it until it is transformed to a stable phase. Many smelters consider arsenic as a penalizing element because its content decreases the value of the concentrate, and in some cases, the value decreases to the point of not being economically viable. Generally, high levels of arsenic are linked with high antimony content, which partially substitutes arsenic, generally finding ores of the form Cu
3(As,Sb)S
4 and Cu
12(As,Sb)
4S
13. The experimental work was carried out with a commercial enargite sample, which had an approximate P
80 of 80 µm; the composition indicated by XRD was 41.5% Cu, 1.8% Fe, 28.5% S, 15.5% As, 0.76% Sb, 360 mg/kg Ag, 9.5 mg/kg Au and the remaining ~12% was composed of silicates, mainly quartz. 10.0 mg of the concentrate was loaded into a laboratory ball mill with 5 balls of 25.4 mm diameter, at room temperature (~25 °C). The atmosphere inside the mill was completely emptied and subsequently filled with argon, air, or oxygen. The Ar and O
2 were treated with an overpressure to prevent air ingress from the outside. In addition, they used a modified mill with heaters, where grinding was carried out at approximately 100 °C.
They performed a computer-simulated evaluation of the thermodynamics in grinding of 10 moles of enargite during its oxidation between 25 and 100 °C, obtaining that there are two main reaction stages; at 25 °C the first stage corresponds to the oxidation to As
2O
3 and CuSO
4 and formation of elemental sulfur; while, the second stage corresponds to the oxidation of sulfur to SO
2, the formation of small amounts of CuS is predicted in the presence of low oxygen concentrations. The formation of As
2O
5 is not considered, since it was not present in the analyzed reports. Obtaining with these data the following reaction, which is highly energetic (−9102 kJ) and will increase the temperature of the system:
They show that at 100 °C the separation of copper and arsenic occurs with less oxygen without the formation of sulfates.
If this reaction were the main reaction, the arsenic could be quickly removed from the system by simple alkaline washing, or by roasting for volatilization.
According to what the authors mentioned, SO
2 can replace oxygen as the oxidizing agent, according to the following reaction, which is only favorable below 35 °C:
The XRD of the experimentally studied enargite concentrate, without grinding, contains mainly enargite and quartz, with minor chalcopyrite and tennantite phases. Milling the concentrate for 50 h under an argon or air atmosphere at room temperature results in a slight modification of its characterization, without the formation of new phases, the peaks obtained by XRD weaken, which is typical of a granulometric redistribution. Working under an oxygen atmosphere at the same temperature, the oxidation starts only 1 h after the beginning of milling and an increase in temperature between 5 and 10 °C was observed, where As2O3 peaks are observed, which increases with increasing milling time. After 10 h CuS was found, and after 50 h CuSO4 × 5H2O. When working the sample in an oxidizing atmosphere and at 100 °C for one hour, As2O3 and tennantite were observed as the main phases. It was concluded that the milling of enargite concentrates is of great help for the preparation of the concentrate, however, it generates high amounts of SO2 emissions. Subsequently, a study on grinding and leaching with 0.5 M HCl was carried out.
After one hour of grinding at room temperature, As2O3 was observed. After 50 h of milling, the enargite and chalcopyrite completely disappeared, and oxidation to CuS, CuSO4, and As2O3 was observed, of which only CuS is insoluble during leaching. After leaching for one hour at 100 °C, it can be observed that all the enargite and chalcopyrite were oxidized, and the predominant phases are tennantite, quartz, and traces of covellite. Finally, they conclude that the leaching of enargite concentrate during milling modifies its reaction mechanisms, in agreement with the reactions initially planned by the authors.
In 1983, Chakraborti et al. [
25] studied the thermodynamics of the Fe-As-S-O system, focusing on the effects of particle size and temperature in order to investigate the effects of different types of atmospheres during the arsenic’s decomposition and oxidation from arsenopyrite. For this purpose, they used natural and synthetic arsenopyrite; the synthetic sample was prepared in the laboratory from iron, arsenic, and sulfur powders 0.0029 inch, which had a purity of 99.99% or higher. The natural sample used came from Gold Hill, Utah; which is associated with large amounts of silica and pyrite. This sample was crushed into +1/8 inch pieces and the pyrite was manually separated. The final sample used contains 20% As comprising FeAsS, along with considerable amounts of pyrite and silica. Under 0.3 g of samples was roasted in different furnaces and atmospheres for periods of 15 to 120 min under oxidizing (O
2), reducing (CO/CO
2), and inert atmospheres (helium). The arsenic volatilizations were immediately transported and condensed for further chemical analysis. The Fe-As-S-O system involves multiple volatile species, the authors state that arsenic can be removed in the gas phase as As
4S
4, As
2S
3, AsS, As
4O
6, As
4, As
2, etc; where the nature of the emission depends on the activities of oxygen, sulfur, and arsenic during roasting. The analysis of this process is complicated due to the lack of thermodynamic information; however, it is possible to represent the equilibrium conditions by means of predominance diagrams, which can be used to evaluate the roasting process. Similar to most gas–solidus reactions, arsenic removal from arsenopyrite increases with increasing temperature. The influence of temperature on the removal kinetics is large, and for particles between 0.0029 and 0.0021 inch it is twice as fast as for particles between 0.0049 and 0.0041 inch; which is a direct consequence of the variation in surface area.
Under an inert atmosphere, arsenopyrite roasting occurs by volatilization of As
4(g), according to the following reaction.
During the process, elemental arsenic is precipitated, and the possibility of the formation of gaseous arsenic sulfides is ruled out. However, an inert atmosphere does not ensure the decomposition of arsenopyrite with the formation of FeS and As4, because sulfides present in the initial sample, such as FeS, can cause a high S2(g) pressure, which can cause arsenic to be trapped in a liquid As-S mixture, which would not allow arsenic to escape to the vapor phase.
Under a reducing atmosphere, the rate and removal of arsenic are increased compared with an inert atmosphere. By adding CO
(g) to a neutral atmosphere, the kinetics is enhanced, however, by adding CO
2(g) the kinetics decreases. The addition of CO
(g) and CO
2(g) to an inert atmosphere generates the formation of arsenic sulfide vapors and the formation of Fe
3O
4. Therefore, it is concluded that the slight presence of oxygen in the atmosphere volatilizes arsenic sulfides to the gas phase, such as AsS, As
4S
4, and As
2S
3, according to the following reactions:
Under oxidizing atmosphere, at high O2(g) pressures arsenic can be retained in the solid products by the formation of FeAsO4 or As2O5. At oxygen pressures of PO2 = 10–21 atm, the predominant arsenic species in the gas phase was As4; whereas at PO2 > 10–21 atm, the predominant arsenic species in the gas phase was As4O6.
The authors conclude that an increase in temperature increases the percentage of arsenic removal. Furthermore, inert, reducing and oxidizing atmospheres can be used for arsenic removal. In an inert atmosphere, the main volatilizations are As4, As2, and S2. A reducing atmosphere favors the formation of arsenic sulfides in the gas phase; and an oxidizing atmosphere favors the volatilization as As4O6.
In 1988, Secco et al. [
55] studied the decomposition of enargite in a neutral and oxidizing atmosphere with the objective of determining the behavior of enargite during its decomposition and oxidation. The most commonly used methods for arsenic removal from enargite concentrates are roasting and smelting where almost all the arsenic is removed in the gas phase. For the study, they used natural enargite from Saint Joe Gold Company’s El Indio mine, which contained 95% enargite, 1.6% pyrite, 1.1% quartz, and 2.3% minor elements. Since both neutral (N
2) and oxidizing (air) atmospheres are used industrially, both were studied. The equipment used was a Setaram DTA. The samples were analyzed by XRD because they were too small to be analyzed chemically. 2–3 g of sample were heated at a rate of 10 °C/h, the study temperatures were 500, 550, 600, and 700 °C, with an inlet gas flow of 0.1–1.0 L/min. The results obtained under a neutral atmosphere indicate that the decomposition of enargite starts at 200 °C and ends at approximately 500 °C, with an endothermic peak at 400 °C. Its decomposition occurs spontaneously at around 600 °C, according to the following reaction:
For 200 min of roasting, air flows at 1 L/min, arsenic removal of 92.5, 99.0, and 99.9% is obtained at 500, 550, and 600 °C, respectively. For a flow rate of 0.1 L/min, a removal of 89.4% is obtained for 500 °C, and 97.8% for 600 °C. Arsenic retention in the calcine increases with the presence of copper and iron oxides, due to the formation of arsenates.
The results obtained for a pure oxygen atmosphere show similarity with an endothermic peak at 400 °C. According to the literature, volatilized sulfur should be extracted as SO
2 and arsenic volatilizes as As
2O
3, according to the following reaction:
In addition, it is possible for elemental sulfur to be oxidized and arsenic to be volatilized as sulfur, according to:
However, both reactions are exothermic and do not correspond to the recorded endothermic peak. Therefore, the same decomposition occurs as in an inert atmosphere.
Subsequently, a phase relation analysis is performed for the Cu-As-S system. In the case of the binary Cu-S system, the possible compounds were CuxS, with x = 1, 1.75–1.79, 1.97, and 2, while for the Cu-As system the compounds were CuyAs, with y = 2.375, 2.7–3.0; Cu4As3, Cu4As2, and Cu8-zAs, with 0 < z < 2.8. In the As-S system, three sulfides As2S3, AsS, and As4S3 were found. On the other hand, in the Cu-As-S system Cu3AsS4, Cu12As4S13, Cu6As4S9, and CuAsS were found. Reagent grade Cu, As and S were used for the experimental development of this analysis; the tests were carried out in a vacuum-sealed furnace, at temperatures between 300 to 600 °C, with a variation of 50 °C, for 2 to 35 days, and subsequently analyzed by XRD.
The authors only report the results at 600 °C, where they obtained two stable compounds tennantite and enargite, together with CuyAs, with y = 2.375, 2.7–3.0, Cu8-zAs, with 0 < z < 2.8, CuS and AsS; two liquid phases were found, one for the Cu-As system, and another for the As-S with dissolved copper sulfides, which indicates that the arsenic volatilizations coming from enargite must pass through a molten phase.
In 2007, Mihajlovic et al. [
56] studied the removal of an enargite concentrate by hydrometallurgical treatment prior to roasting, intending to propose a method for arsenic removal from copper concentrates, which consists of a hydrometallurgical treatment to dissolve arsenic with sodium hypochlorite under alkaline conditions from copper concentrates prior to pyrometallurgical treatment. For this purpose, they used enargite crystals from the Bor copper mine, Serbia. It contained 26.25% Cu, 10.34% As, 19.48% S, 3.18% Al
2O
3, 38.12% SiO
2, and minor elements.
Leaching was carried out at a concentration of 0.3 mol/dm3 NaClO using 800 cm3 for 0.5 g of the sample between 25 and 60 °C for 120 min. Isothermal roasting tests were carried out in an electric resistance furnace with thermostatic control between 400 and 800 °C. The progress of the reaction was determined by ICP spectrometry. The solid residues were analyzed by XRD.
During pyrometallurgical tests, when the temperature in enargite roasting exceeds 193 °C, its reaction is described according to:
In the range 193 to 550 °C, part of the As
2O
3 is oxidized to As
2O
5, which subsequently dissociates as gases in the form of As
4O
6 and O
2, yielding the following general reaction:
Finally, they propose that, for the leaching tests, the following mechanism is obtained:
where arsenic can be stabilized by precipitating it by the following reaction.
In 2012, Padilla et al. [
57] studied the mechanisms and kinetics of enargite oxidation reaction at roasting temperatures in order to understand the behavior of enargite under oxidizing atmosphere. The authors emphasize that although the thermal decomposition of enargite under inert atmosphere is well established in the literature, the literature does not present conclusive data for its decomposition under oxidizing atmosphere. The authors performed tests by measuring mass loss in a thermogravimetric analysis equipment, which consists of a vertical furnace with controlled temperature and a microbalance, in which a crucible with approximately 50 mg of sample is suspended through a quartz chain; the atmospheres used were a mixture of nitrogen and oxygen. The experimental work was carried out with pure enargite crystals from the El Indio mine, Barrick Corporation Chile; taken to a particle size distribution between −75 and +53 µm. The mineralogical composition included enargite, covellite, pyrite, and other impurities. The sample used contained 18.6% As, 46.9% Cu, and 33.0% S, which is close to the theoretical composition of the enargite 19.03% As, 48.4% Cu, and 32.5% S.
Preliminary studies were carried out for the oxidation of enargite in order to analyze its decomposition behavior under atmospheres with different oxygen concentrations. It can be observed that the behavior is similar among the weight loss curves, where each one of them is composed of three segments with different lengths and teeth: which would indicate that the oxidation of the enargite would be carried out through three stages, the first two associated with a mass loss, and the last one associated with a mass increase.
Since the first stage corresponds to a rapid weight loss, it is possible that it is affected by mass transfer. To determine this effect, the flow rate of the oxygen-nitrogen mixture injected into the system was varied from 0.6 to 1.5 L/min. The results of these tests indicated that the sample is not affected by mass transfer.
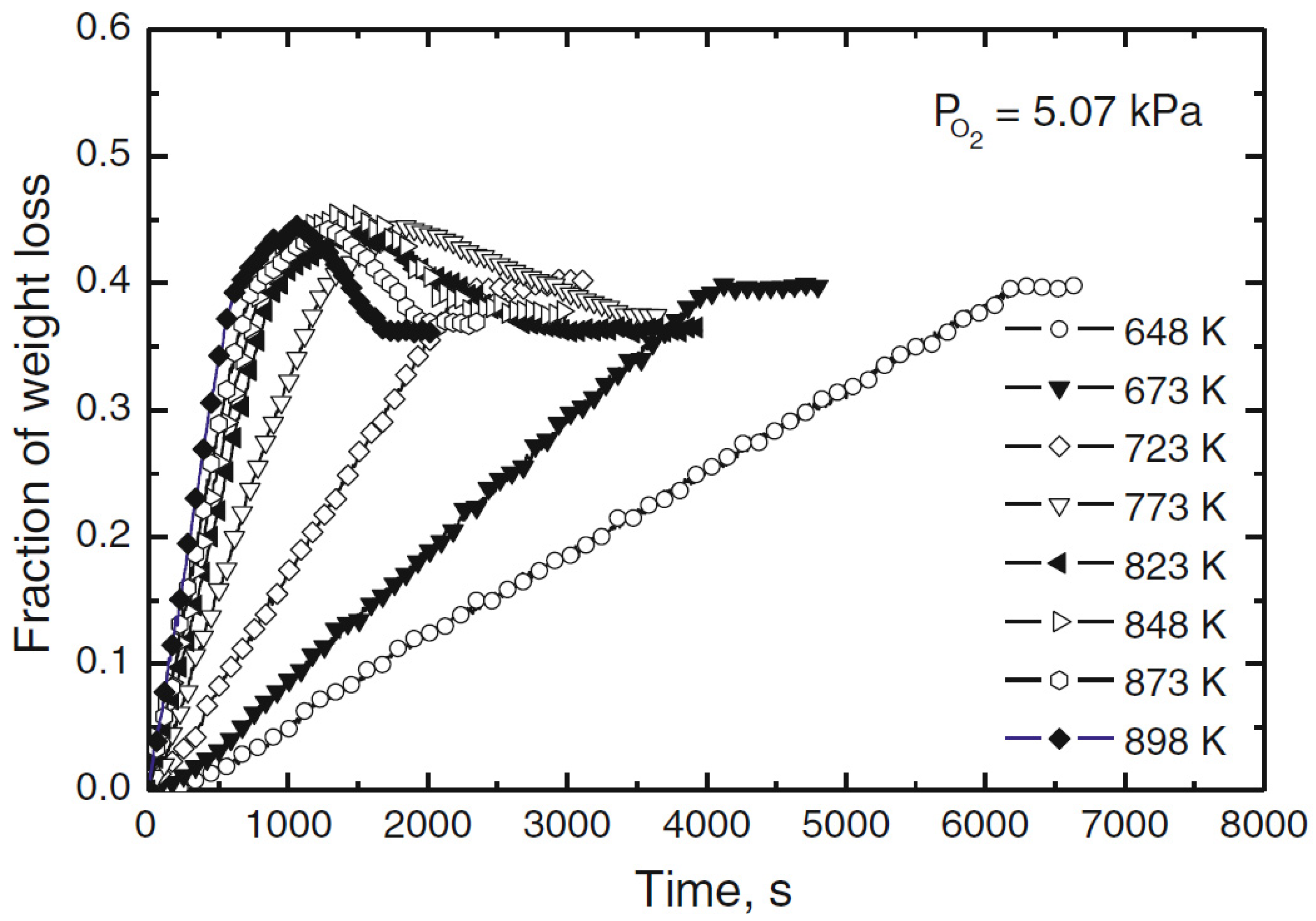
The authors subsequently studied the effect of temperature on the oxidation of enargite in the temperature range between 375 and 625 °C, in order to avoid the formation of molten phases, since the melting points of enargite and tennantite are 687 and 657 °C, respectively. The results obtained are presented in
Figure 6, where it can be seen that as the temperature increases, the speed at which the three stages of decomposition occur increases. All the experiments end with a mass loss fraction of 0.45. At temperatures below 450 °C the occurrence of the second and third stages was observed, and their weight loss fraction is close to 0.4. In addition, the effect of oxygen concentration at 500 °C was studied, from which it was possible to conclude that increasing oxygen concentration causes an increase in the rate of mass loss.
To determine the reaction mechanisms and intermediate compounds, partially reacted samples were analyzed by XRD. A sample was studied at 600 °C, with a partial pressure of oxygen of 1.01 kPa and reacted for 150 and 1400 s, which would correspond to the first stage of oxidation. The sample treated for 150 s presented enargite, tennantite, and slight chalcocite peaks in its composition, whereas the sample treated for 1400 s presented tennantite and chalcocite. Another sample was studied at 600 °C, for 900 and 2700 s, under an oxygen partial pressure of 1.01 and 21.3 kPa, respectively. The 900 s sample, which would correspond to the second oxidation stage, obtained Cu2S and Cu2O, while after 2700 s, CuO and CuO × CuSO4, corresponding to the third stage, were found.
These results indicate that enargite is first oxidized to tennantite as an intermediate compound, followed by the formation of chalcocite. The lack of arsenic compounds indicates that arsenic is volatilized in the first stage in the form of As4O6. In the second stage, chalcocite is oxidized to cuprite, and finally, in the third stage it is oxidized to tenorite, and traces of sulfate are formed, which would explain the mass gain recorded for this stage.
Finally, they study the reaction kinetics of the first stage, since this occurs in a linear way, therefore, it would correspond to a reaction with a constant reaction rate. For this purpose, they analyze the mass loss at different temperatures and a partial pressure of oxygen of 5.07 kPa between 375 and 625 °C, obtaining that the activation energy in the temperature range studied is 44 kJ/mol.
Finally, concluding that the thermal decomposition of enargite occurs through the following three stages.
With formation of sulfate traces.
Finally, the postulated global reaction is:
For the case of the compound arsenic sulfides, we can appreciate the same trend shown in
Figure 1, and previous articles regarding the predominant phase, As
2O
3(g). Therefore, the predominant phase for volatilizations from compound arsenic sulfides is As
2O
3(g), in the form of its dimer As
4O
6(g).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





