

Hydrogen Insertion into Complex-Phase High-Strength Steel during Atmospheric Corrosion at Low Relative Humidity

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Scanning Kelvin Probe
2.2. Hydrogen Content
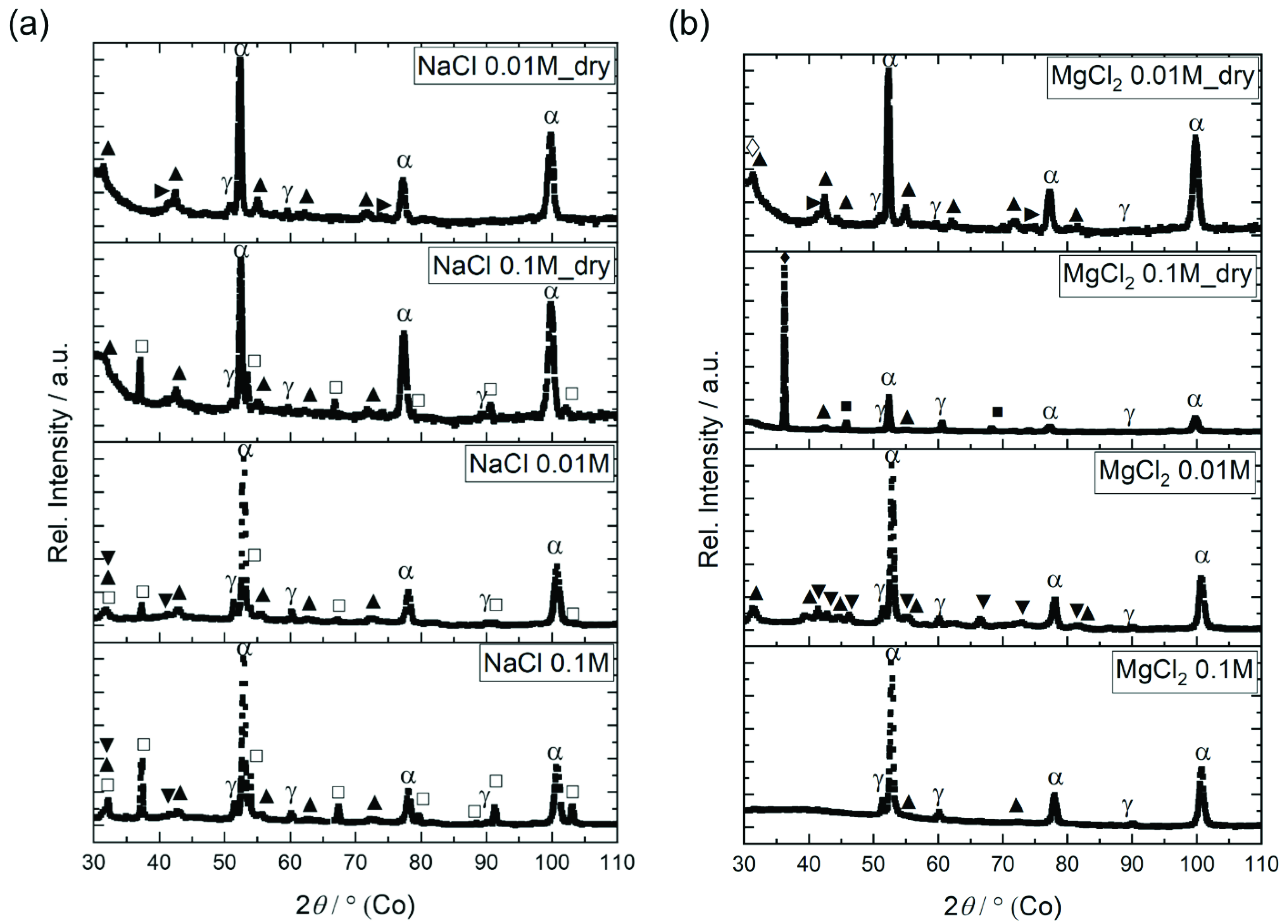
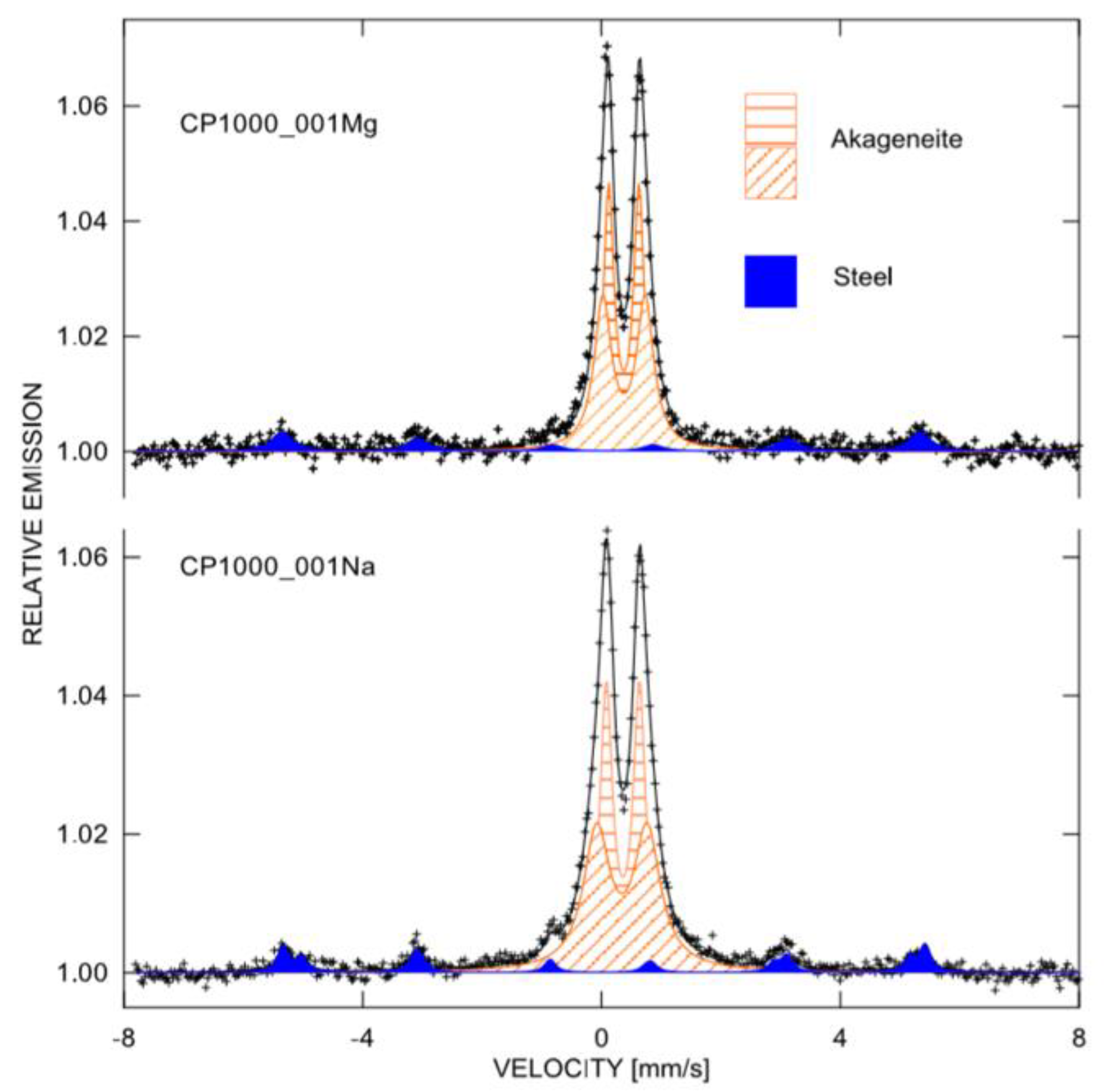
2.3. Corrosion Product Analysis
3. Results
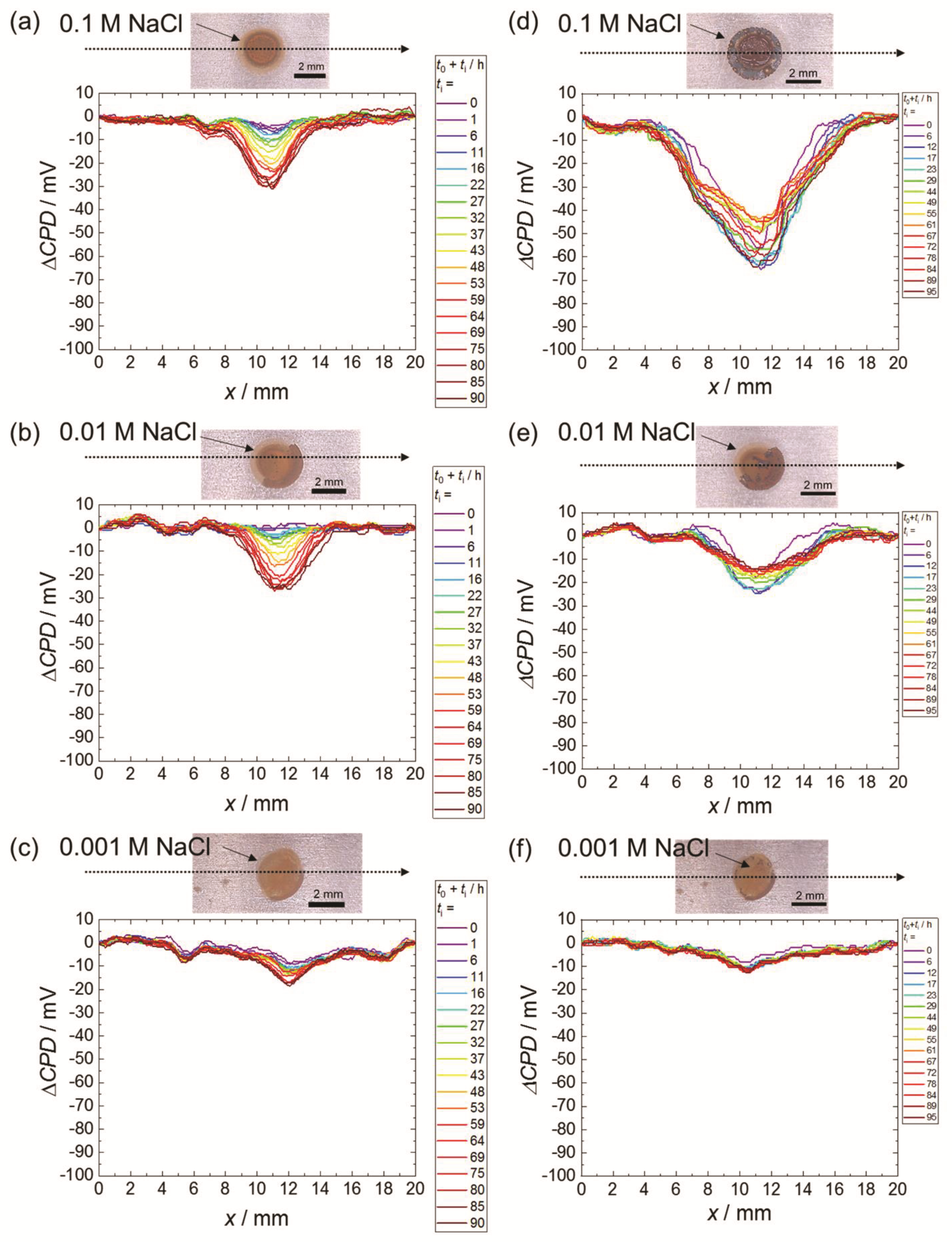
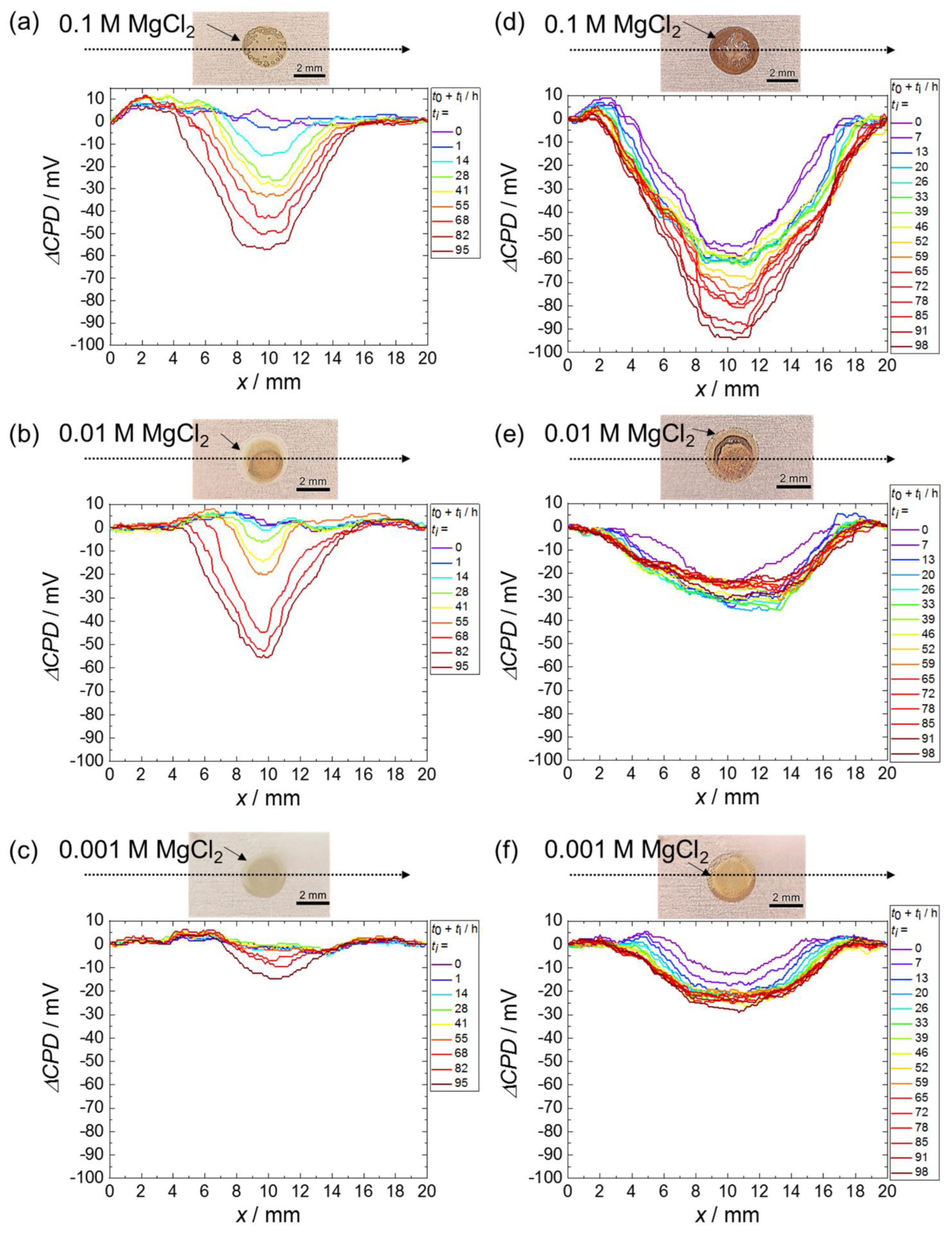
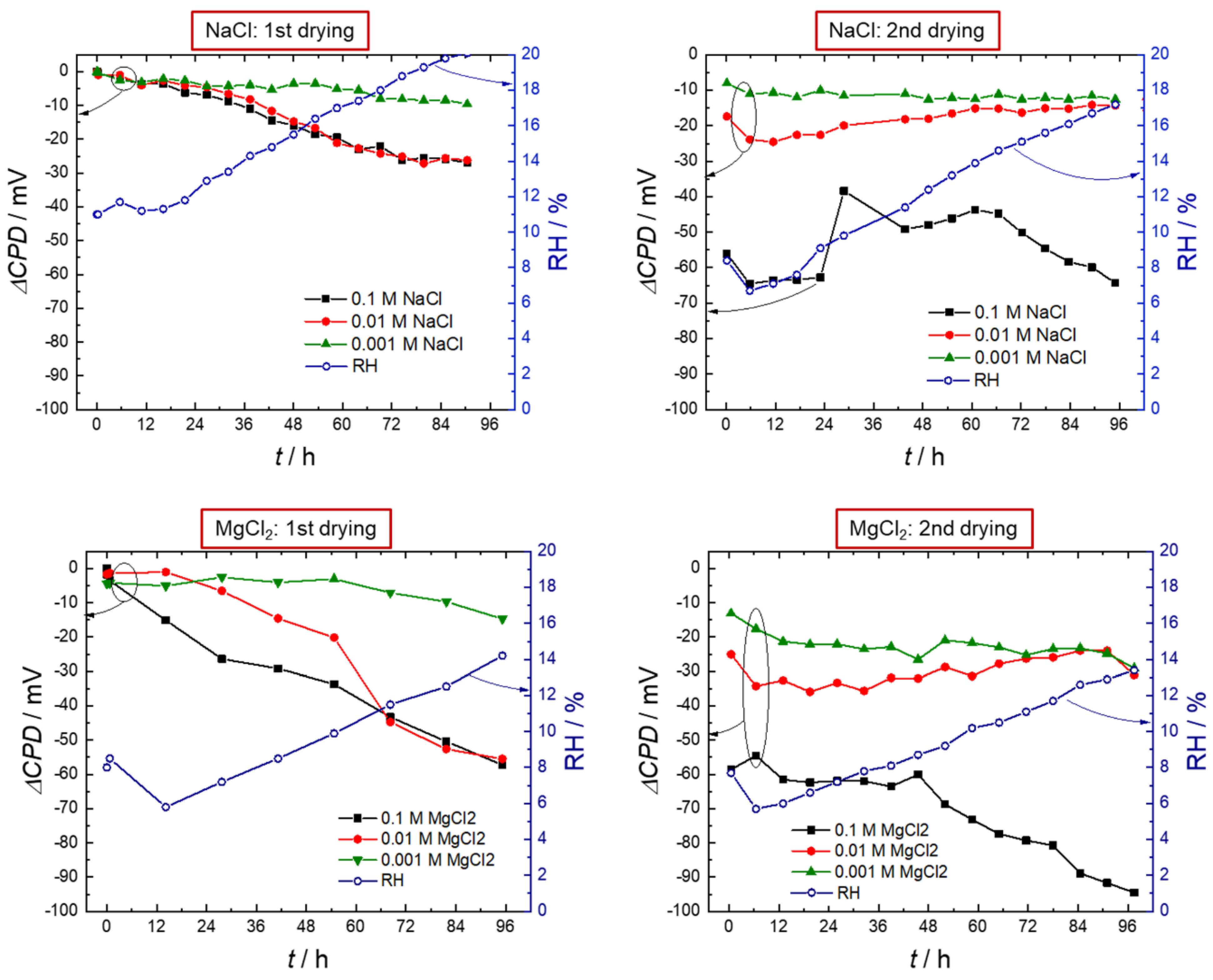
3.1. Scanning Kelvin Probe
3.2. Hydrogen Content
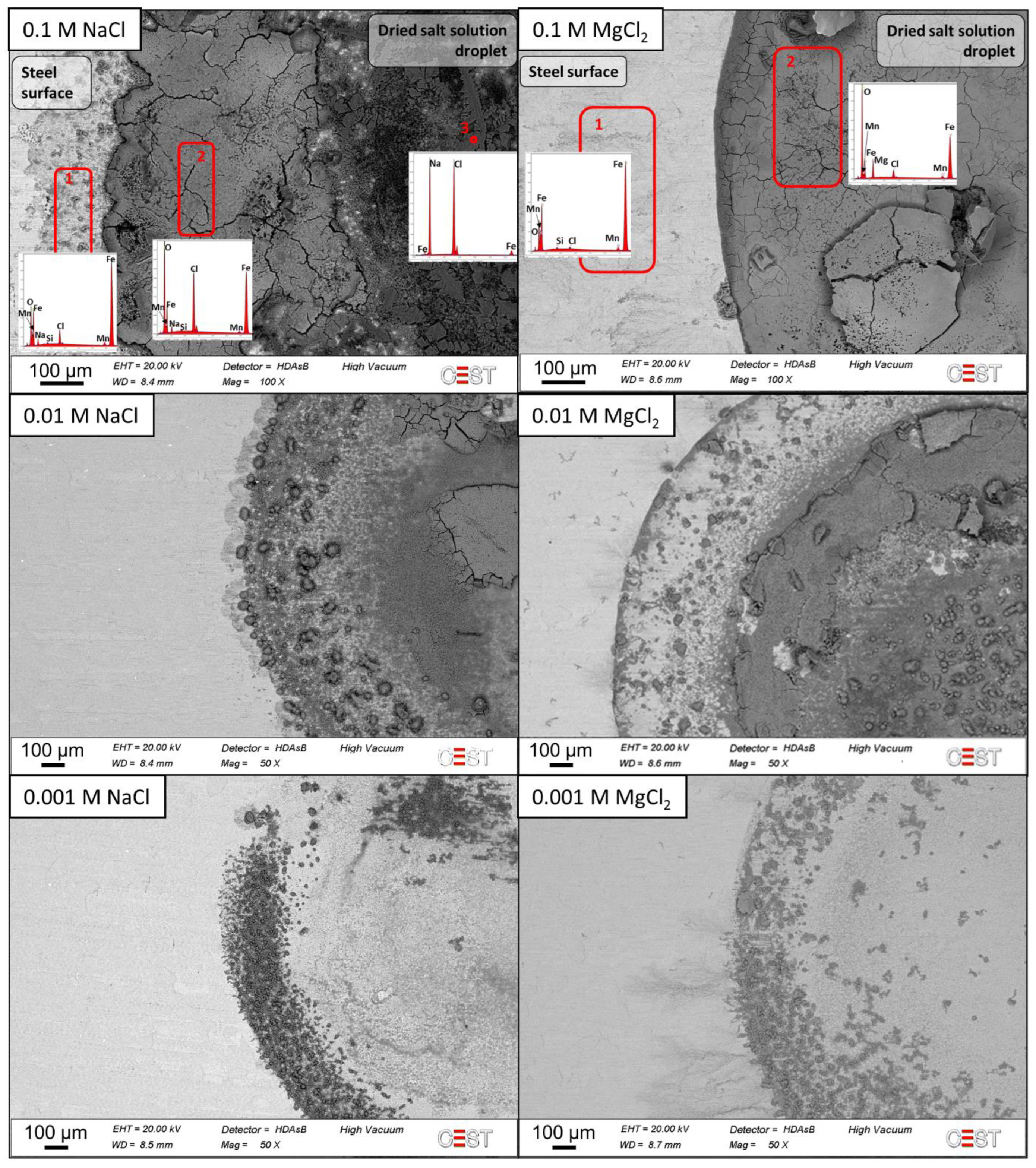
3.3. Corrosion Product Analyses
4. Discussion
5. Conclusions
- (1)
- The hygroscopic behavior of MgCl2 prolonged hydrogen entry during drying down to low levels of relative humidity (>20% RH), which is well below the deliquescence humidity of the salt.
- (2)
- The surface pH below a salt crust of MgCl2 was lower. The crust also slowed down water evaporation and oxygen diffusion.
- (3)
- Iron chlorides detected on MgCl2-contaminated samples enhanced active corrosion under the deposits.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rudomilova, D.; Prošek, T.; Traxler, I.; Faderl, J.; Luckeneder, G.; Schimo-Aichhorn, G.; Muhr, A. Critical Assessment of the Effect of Atmospheric Corrosion Induced Hydrogen on Mechanical Properties of Advanced High Strength Steel. Metals 2020, 11, 44. [Google Scholar] [CrossRef]
- Barrera, O. Understanding and Mitigating Hydrogen Embrittlement of Steels: A Review of Experimental, Modelling and Design Progress from Atomistic to Continuum. J. Mater. Sci. 2018, 53, 6251–6290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehrl, J.; Mraczek, K.; Pichler, A.; Werner, E. Mechanical Properties and Fracture Behavior of Hydrogen Charged AHSS/UHSS Grades at High- and Low Strain Rate Tests. Mater. Sci. Eng. A 2014, 590, 360–367. [Google Scholar] [CrossRef]
- Scharf, R.; Muhr, A.; Luckeneder, G.; Larour, P.; Mraczek, K.; Rehrl, J.; Leomann, F.; Stellnberger, K.-H.; Faderl, J.; Mori, G. Hydrogen Embrittlement of DP-1000 Flat Steel Sheet: Influence of Mechanical Properties, Specimen Geometry, Pre-Damaging and Electrolytically Zinc Galvanizing: Hydrogen Embrittlement of DP-1000 Flat Steel Sheet. Mater. Corros. 2016, 67, 239–250. [Google Scholar] [CrossRef]
- Evers, S.; Senoz, C.; Rohwerder, M. Hydrogen Detection in Metals: A Review and Introduction of a Kelvin Probe Approach. Sci. Technol. Adv. Mater. 2013, 14, 014201. [Google Scholar] [CrossRef]
- Evers, S. The Hydrogen Electrode in the “Dry”: A Kelvin Probe Approach to Measuring Hydrogen in Metals. Electrochem. Commun. 2012, 24, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Schimo, G.; Burgstaller, W.; Hassel, A.W. Potentiodynamic Hydrogen Permeation on Palladium-Kelvin Probe Compared to 3D Printed Microelectrochemical Cell. Electrochem. Commun. 2015, 60, 208–211. [Google Scholar] [CrossRef]
- Schimo, G.; Burgstaller, W.; Hassel, A.W. Influence of Atmospheric Oxygen on Hydrogen Detection on Pd Using Kelvin Probe Technique. J. Solid State Electrochem. 2018, 22, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-H.; Krieger, W.; Rohwerder, M. On the Robustness of the Kelvin Probe Based Potentiometric Hydrogen Electrode Method and Its Application in Characterizing Effective Hydrogen Activity in Metal: 5 wt. % Ni Cold-Rolled Ferritic Steel as an Example. Sci. Technol. Adv. Mater. 2019, 20, 1073–1089. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.; McMurray, H.N.; Newman, R.C. Surface Oxide Reduction by Hydrogen Permeation through Iron Foil Detected Using a Scanning Kelvin Probe. Electrochem. Commun. 2013, 27, 144–147. [Google Scholar] [CrossRef]
- Schaller, R.F.; Scully, J.R. Measurement of Effective Hydrogen Diffusivity Using the Scanning Kelvin Probe. Electrochem. Commun. 2014, 40, 42–44. [Google Scholar] [CrossRef]
- Schimo, G.; Burgstaller, W.; Hassel, A.W. Rolling Direction Dependent Diffusion Coefficients of Hydrogen in Ferritic Steel by SDCM Charging and SKP Probing. ISIJ Int. 2016, 56, 487–491. [Google Scholar] [CrossRef] [Green Version]
- Nazarov, A.P.; Marshakov, A.I.; Rybkina, A.A. Iron Hydrogenation under Atmospheric Corrosion. Studies Using a Scanning Vibrating Microscope. Prot. Met. Phys. Chem. Surf. 2015, 51, 347–359. [Google Scholar] [CrossRef]
- Nazarov, A.; Vucko, F.; Thierry, D. Scanning Kelvin Probe for Detection of the Hydrogen Induced by Atmospheric Corrosion of Ultra-High Strength Steel. Electrochim. Acta 2016, 216, 130–139. [Google Scholar] [CrossRef]
- Nazarov, A.; Vucko, F.; Thierry, D. Scanning Kelvin Probe Investigation of High-Strength Steel Surface after Impact of Hydrogen and Tensile Strain. CMD 2020, 1, 187–197. [Google Scholar] [CrossRef]
- Evans, U.R. The Ferroxyl Indicator in Corrosion Research, With Special Reference to the Controversy Regarding the Cause of Pitting. Met. Ind. 1926, 11, 481–508. [Google Scholar]
- Soulié, V.; Lequien, F.; Ferreira-Gomes, F.; Moine, G.; Feron, D.; Prene, P.; Moehwald, H.; Zemb, T.; Riegler, H. Salt-Induced Iron Corrosion under Evaporating Sessile Droplets of Aqueous Sodium Chloride Solutions. Mater. Corros. 2017, 68, 927–934. [Google Scholar] [CrossRef]
- Risteen, B.E.; Schindelholz, E.; Kelly, R.G. Marine Aerosol Drop Size Effects on the Corrosion Behavior of Plain Carbon Steel. ECS Trans. 2014, 58, 1–12. [Google Scholar] [CrossRef]
- Katona, R.M.; Tokuda, S.; Perry, J.; Kelly, R.G. Design, Construction, and Validation for in-Situ Water Layer Thickness Determination during Accelerated Corrosion Testing. Corros. Sci. 2020, 175, 108849. [Google Scholar] [CrossRef]
- Katona, R.M.; Kelly, R.G.; Bryan, C.R.; Schaller, R.F.; Knight, A.W. Use of in Situ Raman Spectroelectrochemical Technique to Explore Atmospheric Corrosion in Marine-Relevant Environments. Electrochem. Commun. 2020, 118, 106768. [Google Scholar] [CrossRef]
- Strebl, M.; Bruns, M.; Virtanen, S. Editors’ Choice—Respirometric in Situ Methods for Real-Time Monitoring of Corrosion Rates: Part, I. Atmospheric Corrosion. J. Electrochem. Soc. 2020, 167, 021510. [Google Scholar] [CrossRef]
- Rudomilova, D.; Prošek, T.; Luckeneder, G. Techniques for Investigation of Hydrogen Embrittlement of Advanced High Strength Steels. Corros. Rev. 2018, 36, 413–434. [Google Scholar] [CrossRef]
- Schindelholz, E.; Kelly, R.G.; Cole, I.S.; Ganther, W.D.; Muster, T.H. Comparability and Accuracy of Time of Wetness Sensing Methods Relevant for Atmospheric Corrosion. Corros. Sci. 2013, 67, 233–241. [Google Scholar] [CrossRef]
- Schindelholz, E.; Tsui, L.; Kelly, R.G. Hygroscopic Particle Behavior Studied by Interdigitated Array Microelectrode Impedance Sensors. J. Phys. Chem. A 2014, 118, 167–177. [Google Scholar] [CrossRef]
- Stratmann, M.; Streckel, H. On the Atmospheric Corrosion of Metals Which Are Covered with Thin Electrolyte Layers—I. Verification of the Experimental Technique. Corros. Sci. 1990, 30, 681–696. [Google Scholar] [CrossRef]
- Stratmann, M.; Streckel, H. On the Atmospheric Corrosion of Metals Which Are Covered with Thin Electrolyte Layers—II. Experimental Results. Corros. Sci. 1990, 30, 697–714. [Google Scholar] [CrossRef]
- Schindelholz, E.; Risteen, B.E.; Kelly, R.G. Effect of Relative Humidity on Corrosion of Steel under Sea Salt Aerosol Proxies: I. NaCl. J. Electrochem. Soc. 2014, 161, C450–C459. [Google Scholar] [CrossRef]
- Schindelholz, E.; Risteen, B.E.; Kelly, R.G. Effect of Relative Humidity on Corrosion of Steel under Sea Salt Aerosol Proxies II. MgCl2, Artificial Seawater. J. Electrochem. Soc. 2014, 161, C460–C470. [Google Scholar] [CrossRef]
- Greenspan, L. Humidity Fixed Points of Binary Saturated Aqueous Solutions. J. Res. Natl. Bur. Stand 1977, 81A, 89–96. [Google Scholar] [CrossRef]
- Huang, L.; Li, Z.; Lou, Y.; Cao, F.; Zhang, D.; Li, X. Recent Advances in Scanning Electrochemical Microscopy for Biological Applications. Materials 2018, 11, 1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, E.; Matsukado, K.; Wang, M.; Tsuzaki, K. Evaluation of Hydrogen Entry into High Strength Steel under Atmospheric Corrosion. Corros. Sci. 2010, 52, 2758–2765. [Google Scholar] [CrossRef]
- Akiyama, E. Hydrogen Entry into Fe and High Strength Steels under Simulated Atmospheric Corrosion. Electrochim. Acta 2011, 56, 1799–1805. [Google Scholar] [CrossRef]
- Omura, T.; Kudo, T.; Fujimoto, S. Environmental Factors Affecting Hydrogen Entry into High Strength Steel Due to Atmospheric Corrosion. Mater. Trans. 2006, 12, 2956–2962. [Google Scholar] [CrossRef] [Green Version]
- Ootsuka, S.; Fujita, S.; Tada, E.; Nishikata, A.; Tsuru, T. Evaluation of Hydrogen Absorption into Steel in Automobile Moving Environments. Corros. Sci. 2015, 98, 430–437. [Google Scholar] [CrossRef]
- Traxler, I.; Schimo-Aichhorn, G.; Muhr, A.; Commenda, C.; Jerrar, A.; Sagl, R.; Mraczek, K.; Rudomilova, D.; Luckeneder, G.; Duchaczek, H.; et al. Optimization of Metallographic Sample Preparation for AFM/SKPFM Based Phase Distinction of Complex and Dual Phase High Strength Steels. Pract. Metallogr. 2021, 58, 308–331. [Google Scholar] [CrossRef]
- Rudomilova, D.; Prošek, T.; Salvetr, P.; Knaislová, A.; Novák, P.; Kodým, R.; Schimo-Aichhorn, G.; Muhr, A.; Duchaczek, H.; Luckeneder, G. The Effect of Microstructure on Hydrogen Permeability of High Strength Steels. Mater. Corros. 2019, maco.201911357. [Google Scholar] [CrossRef]
- Schuttlefield, J.D.; Cox, D.; Grassian, V.H. An Investigation of Water Uptake on Clays Minerals Using ATR-FTIR Spectroscopy Coupled with Quartz Crystal Microbalance Measurements. J. Geophys. Res. Atmos. 2007, 112. [Google Scholar] [CrossRef] [Green Version]
- Prosek, T.; Iversen, A.; Taxén, C.; Thierry, D. Low-Temperature Stress Corrosion Cracking of Stainless Steels in the Atmosphere in the Presence of Chloride Deposits. Corrosion 2009, 65, 105–117. [Google Scholar] [CrossRef]
- Rudomilova, D.; Prošek, T.; Ström, M. Hydrogen Entry into Steel Under Corrosion Products. Corrosion 2021, 77, 427–432. [Google Scholar] [CrossRef]
- Stratmann, M.; Müller, J. The Mechanism of the Oxygen Reduction on Rust-Covered Metal Substrates. Corros. Sci. 1994, 36, 327–359. [Google Scholar] [CrossRef]
- Schwertmann, U.; Cornell, R.M. Iron Oxides in the Laboratory: Preparation and Characterization; Wiley: Hoboken, NJ, USA, 2000; ISBN 978-3-527-29669-9. [Google Scholar]
- Katona, R.M.; Knight, A.W.; Schindelholz, E.J.; Bryan, C.R.; Schaller, R.F.; Kelly, R.G. Quantitative Assessment of Environmental Phenomena on Maximum Pit Size Predictions in Marine Environments. Electrochim. Acta 2021, 370, 137696. [Google Scholar] [CrossRef]
- Leygraf, C.; Odnevall Wallinder, I.; Tidblad, J.; Graedel, T.E. Atmospheric Corrosion, 2nd ed.; The Electrochemical Society Series; Wiley: Hoboken, NJ, USA, 2016; ISBN 978-1-118-76227-1. [Google Scholar]
- Tsuru, T.; Huang, Y.; Ali, R.; Nishikata, A. Hydrogen Entry into Steel during Atmospheric Corrosion Process. Corros. Sci. 2005, 47, 1431–2440. [Google Scholar] [CrossRef]
- Wang, Y.; Yamanishi, J.; Haruna, T. Effect of Humidity of Air on Hydrogen Absorption into Fe with Rust Layer Containing MgCl2 during Atmospheric Corrosion. ISIJ Int. 2021, 61, 1143–1150. [Google Scholar] [CrossRef]
- Li, S.; Hihara, L.H. Atmospheric-Corrosion Electrochemistry of NaCl Droplets on Carbon Steel. J. Electrochem. Soc. 2012, 159, C461–C469. [Google Scholar] [CrossRef]
- Turgoose, S. Post-Excavation Changes in Iron Antiquities. Stud. Conserv. 1982, 27, 97–101. [Google Scholar] [CrossRef]
- Watkinson, D.; Lewis, M.T. Desiccated Storage of Chloride-Contaminated Archaeological Iron Objects. Stud. Conserv. 2005, 50, 241–252. [Google Scholar] [CrossRef]
- Wise, M.E.; Martin, S.T.; Russell, L.M.; Buseck, P.R. Water Uptake by NaCl Particles Prior to Deliquescence and the Phase Rule. Aerosol Sci. Technol. 2008, 42, 281–294. [Google Scholar] [CrossRef] [Green Version]
- Bruzewicz, D.A.; Checco, A.; Ocko, B.M.; Lewis, E.R.; McGraw, R.L.; Schwartz, S.E. Reversible Uptake of Water on NaCl Nanoparticles at Relative Humidity below Deliquescence Point Observed by Noncontact Environmental Atomic Force Microscopy. J. Chem. Phys. 2011, 134, 044702. [Google Scholar] [CrossRef] [PubMed]
- Katona, R.M.; Carpenter, J.C.; Knight, A.W.; Bryan, C.R.; Schaller, R.F.; Kelly, R.G.; Schindelholz, E.J. Importance of the Hydrogen Evolution Reaction in Magnesium Chloride Solutions on Stainless Steel. Corros. Sci. 2020, 177, 108935. [Google Scholar] [CrossRef]
- Prosek, T.; Thierry, D.; Taxén, C.; Maixner, J. Effect of Cations on Corrosion of Zinc and Carbon Steel Covered with Chloride Deposits under Atmospheric Conditions. Corros. Sci. 2007, 49, 2676–2693. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmax | Simax | Mnmax | Pmax | Smax | Altotal | Cr + Momax | Ti + Nbmax | Vmax | Bmax |
|---|---|---|---|---|---|---|---|---|---|
| 0.23 | 1.00 | 2.70 | 0.08 | 0.015 | 0.015–2.0 | 1.00 | 0.15 | 0.22 | 0.005 |
| Salt Type and Concentration/mol L−1 | 0.001 | pH Value (22 °C) 0.01 | 0.1 | DRH/% |
|---|---|---|---|---|
| NaCl | 6.8 | 6.7 | 6.6 | 74–76 [23,29,37] |
| MgCl2 | 6.5 | 6.2 | 5.9 | 33–36 [29,38] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schimo-Aichhorn, G.; Traxler, I.; Muhr, A.; Commenda, C.; Rudomilova, D.; Schneeweiss, O.; Luckeneder, G.; Duchaczek, H.; Stellnberger, K.-H.; Faderl, J.; et al. Hydrogen Insertion into Complex-Phase High-Strength Steel during Atmospheric Corrosion at Low Relative Humidity. Metals 2022, 12, 624. https://doi.org/10.3390/met12040624
Schimo-Aichhorn G, Traxler I, Muhr A, Commenda C, Rudomilova D, Schneeweiss O, Luckeneder G, Duchaczek H, Stellnberger K-H, Faderl J, et al. Hydrogen Insertion into Complex-Phase High-Strength Steel during Atmospheric Corrosion at Low Relative Humidity. Metals. 2022; 12(4):624. https://doi.org/10.3390/met12040624
Chicago/Turabian StyleSchimo-Aichhorn, Gabriela, Ines Traxler, Andreas Muhr, Christian Commenda, Darya Rudomilova, Oldřich Schneeweiss, Gerald Luckeneder, Hubert Duchaczek, Karl-Heinz Stellnberger, Josef Faderl, and et al. 2022. "Hydrogen Insertion into Complex-Phase High-Strength Steel during Atmospheric Corrosion at Low Relative Humidity" Metals 12, no. 4: 624. https://doi.org/10.3390/met12040624
APA StyleSchimo-Aichhorn, G., Traxler, I., Muhr, A., Commenda, C., Rudomilova, D., Schneeweiss, O., Luckeneder, G., Duchaczek, H., Stellnberger, K.-H., Faderl, J., Prošek, T., Stifter, D., Hassel, A. W., & Hild, S. (2022). Hydrogen Insertion into Complex-Phase High-Strength Steel during Atmospheric Corrosion at Low Relative Humidity. Metals, 12(4), 624. https://doi.org/10.3390/met12040624