
First-Principles Investigation on Phase Stability, Mechanical Properties, Bonding Characteristic and Slip Properties of Ti-Co Binary Intermetallic Compounds


Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
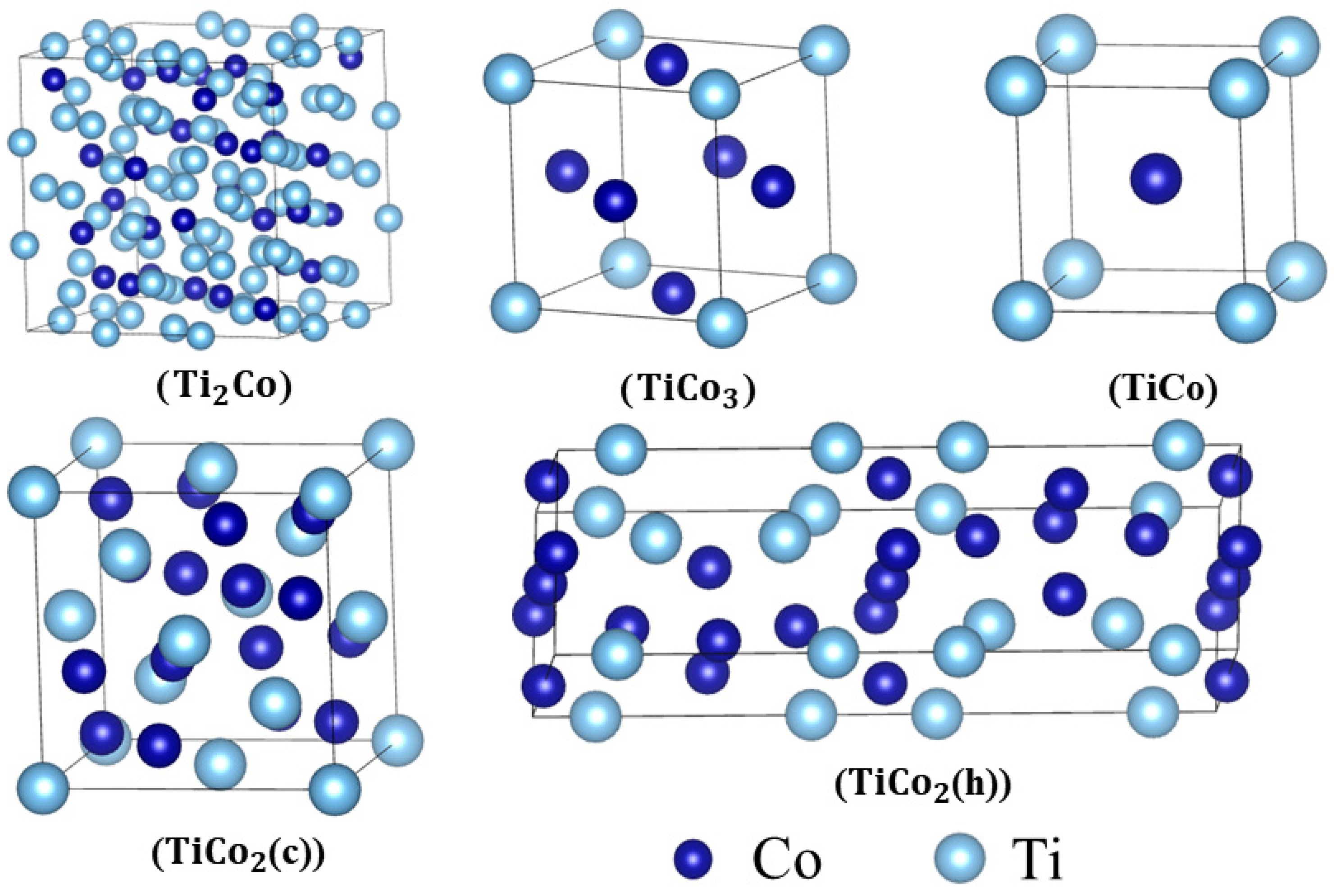
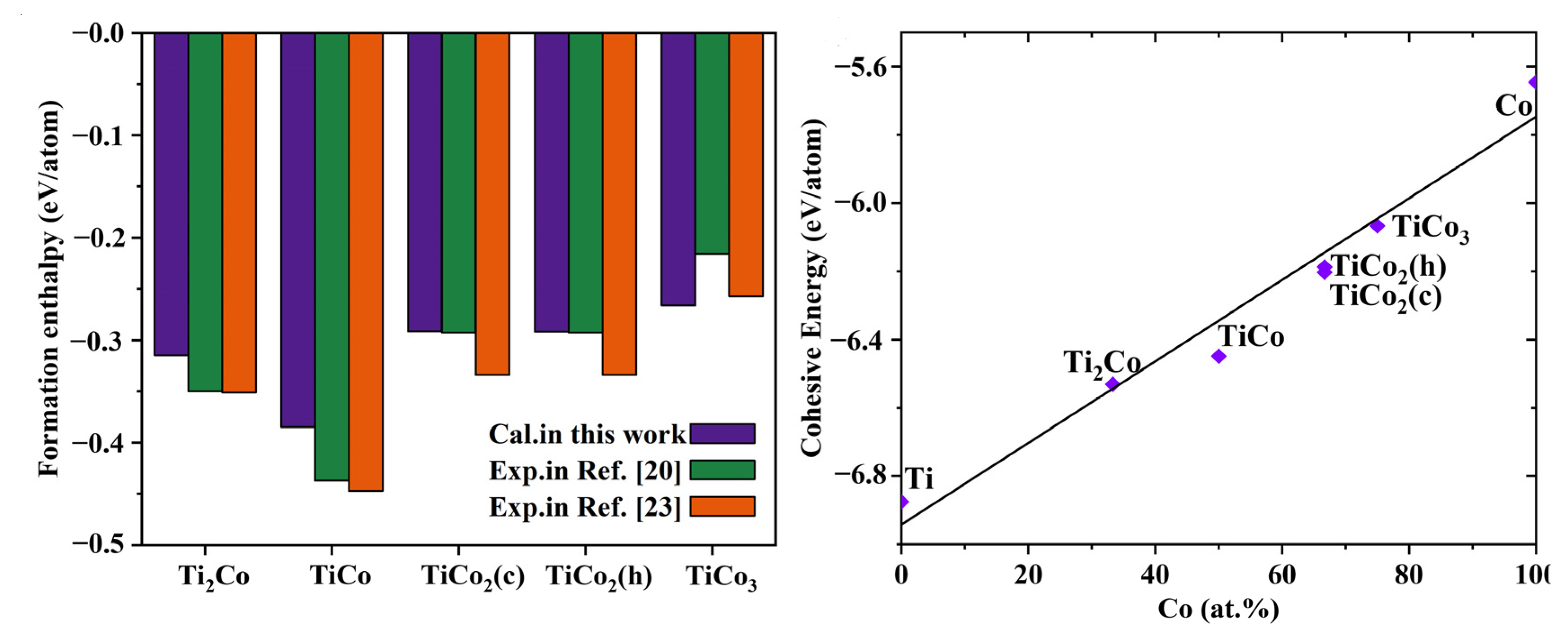
3.1. Lattice Parameters and Phase Stability
3.2. Elastic Properties
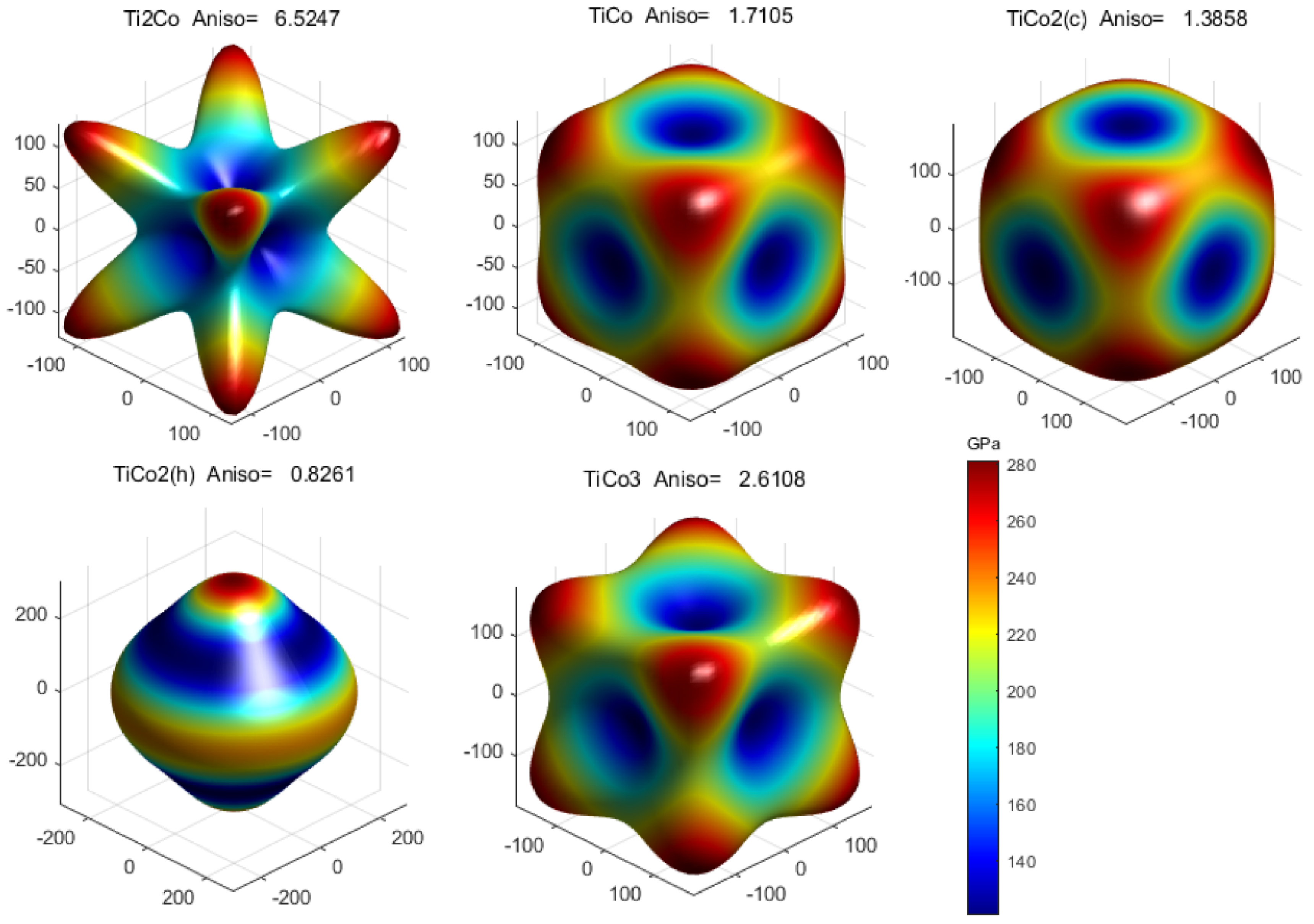
3.3. Mechanical Anisotropy
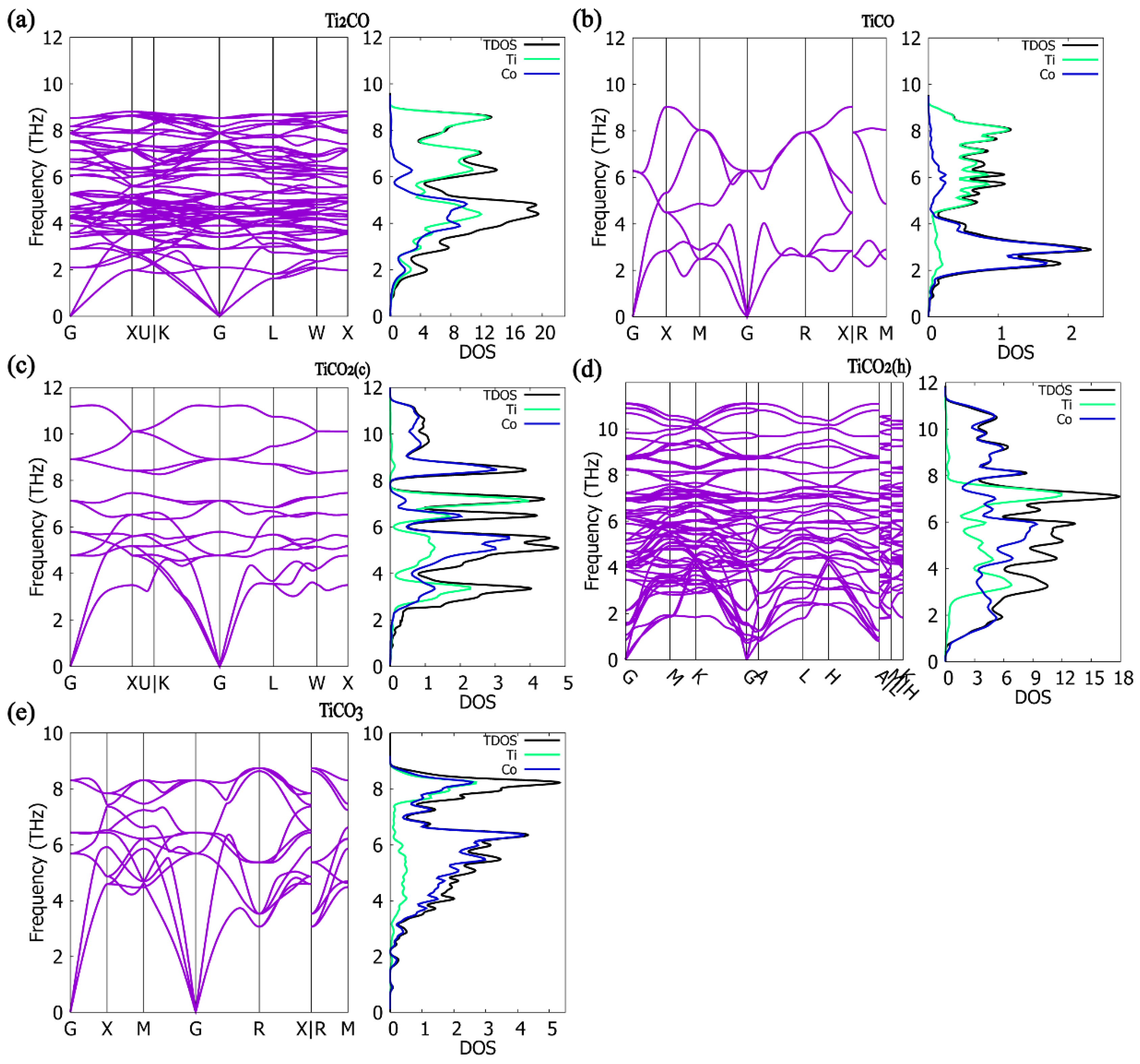
3.4. Phonon Properties
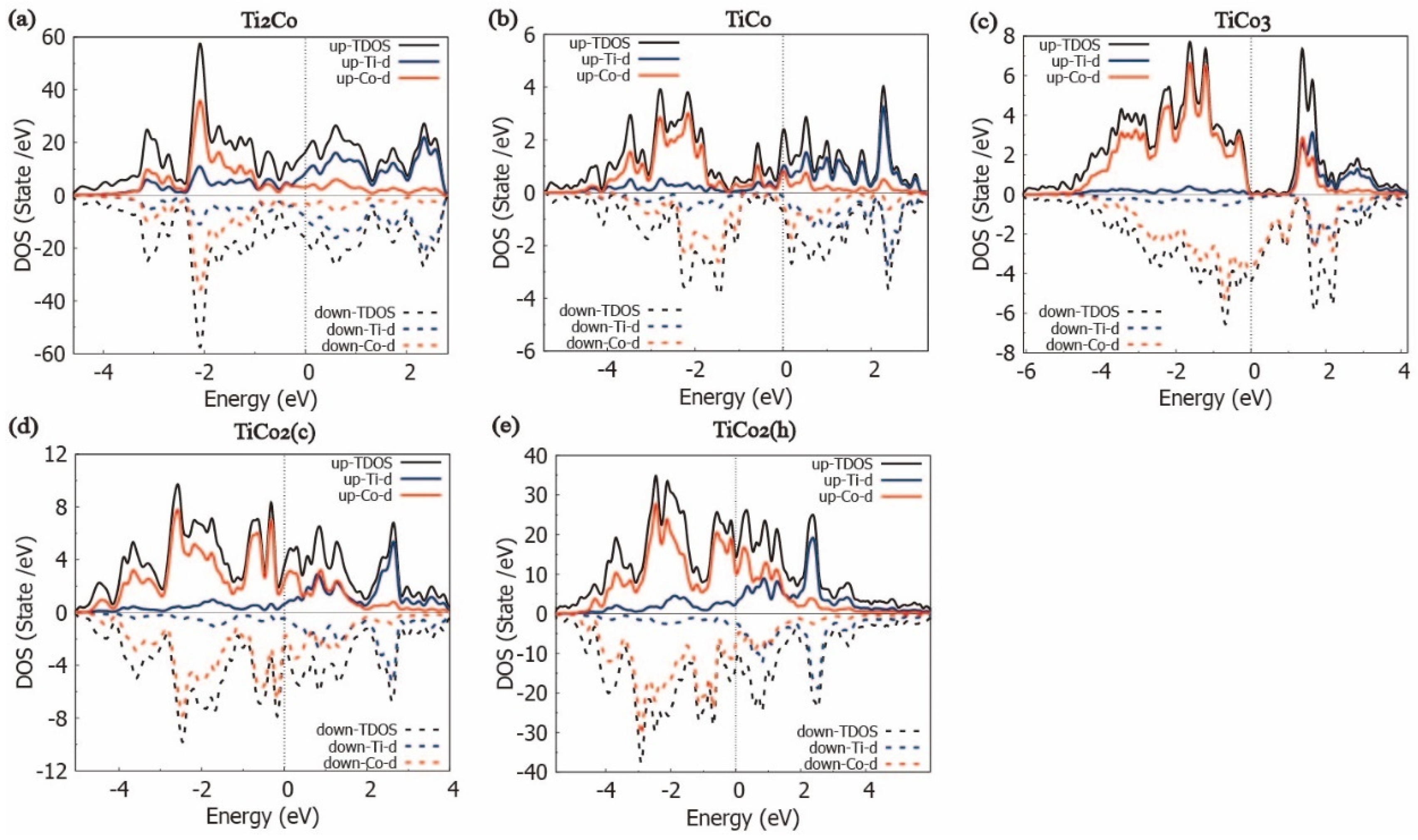
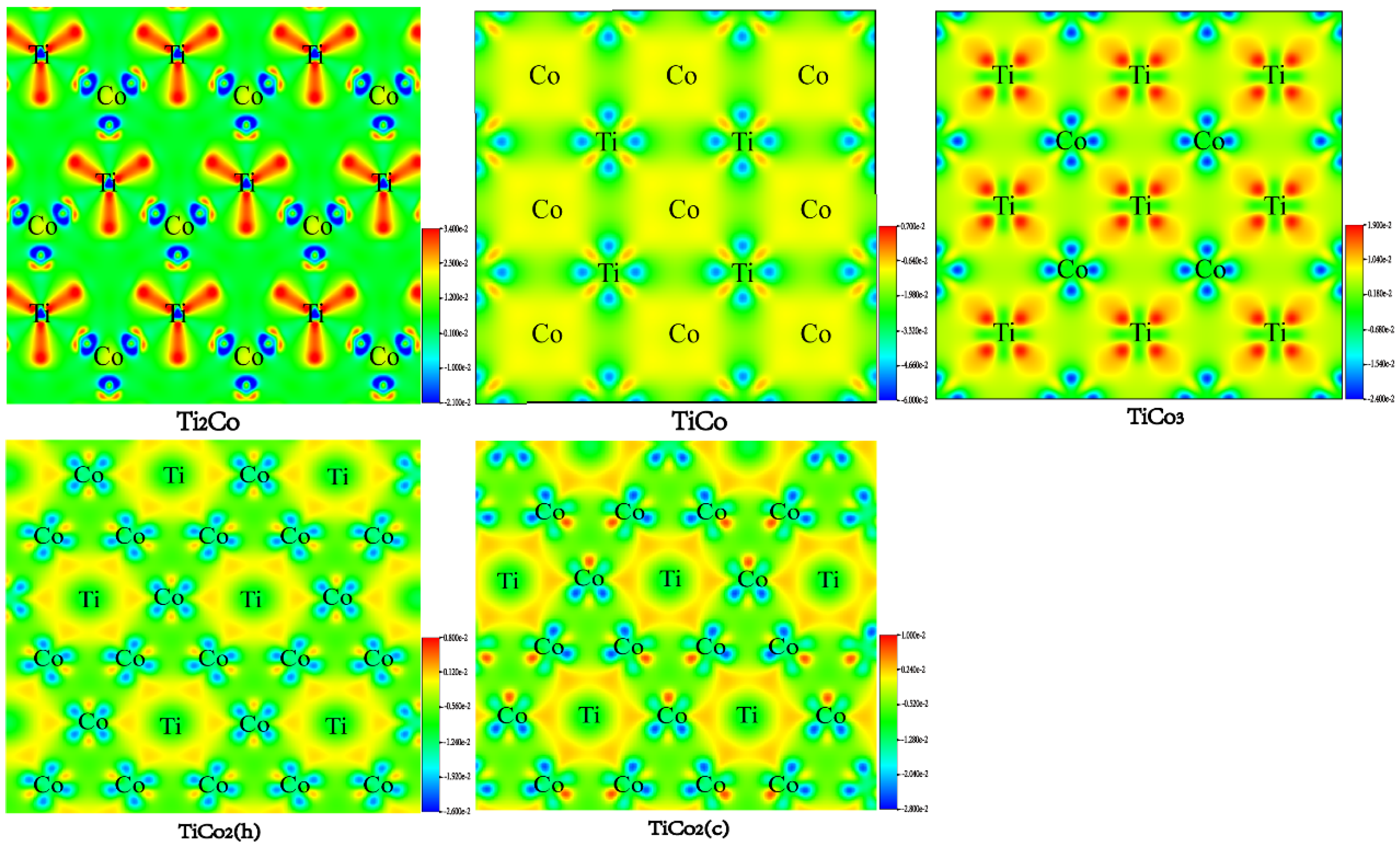
3.5. Bonding Characteristic
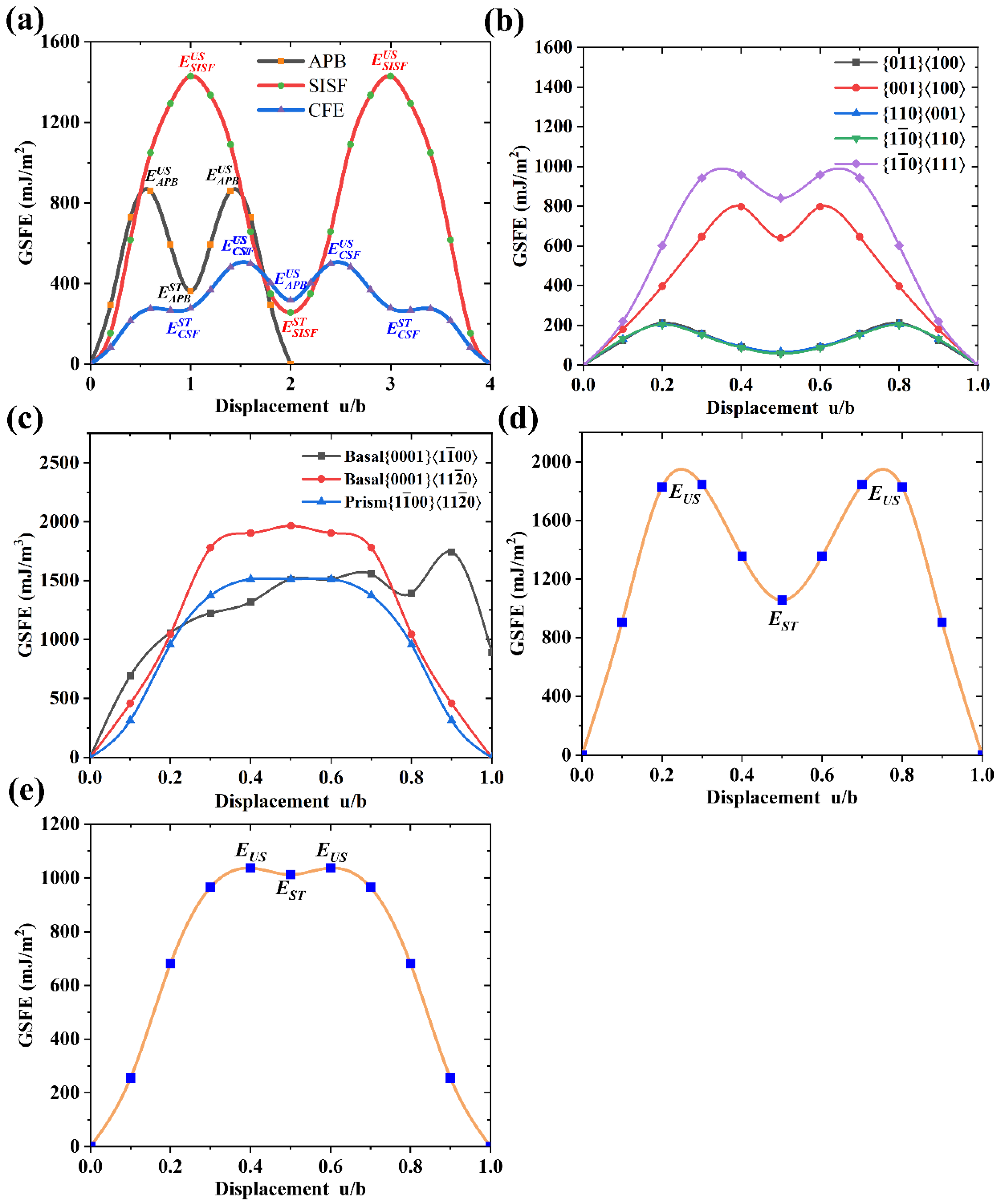
3.6. Slip Properties
4. Conclusions
- (1)
- According to thermodynamic and the elastic standards and phonon properties calculated by the frozen phonon method, the five Ti-Co compounds are thermodynamically stable, and TiCo has the lowest enthalpy of formation and is the most stable phase.
- (2)
- Ti2Co shows the strongest anisotropy, and TiCo2(h) has the weakest anisotropy. The three-dimensional surface of the general anisotropy index and Young’s modulus indicate that the magnitude relationship of mechanical anisotropy is Ti2Co > TiCo3 > TiCo > TiCo2(c) > TiCo2(h).
- (3)
- The calculations of bonding characteristic and differential charge density distributions show that Ti-Co compounds are composed of metallic and covalent-like bonds. TiCo shows stronger covalent-like bonding characteristics. Additionally, TiCo and TiCo2(h) are ferromagnetic, TiCo3 is ferromagnetic, and Ti2Co and TiCo2(c) are non-magnetic.
- (4)
- The calculated stacking fault energy of the slip systems shows that the anomalous ductility of Ti-Co compounds mainly comes from the complex slip systems and the lower slip energy barrier of the compounds.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yeh, C. Preparation of CoTi intermetallics by self-propagating combustion synthesis. J. Alloy. Compd. 2005, 396, 228–232. [Google Scholar] [CrossRef]
- Zhu, Y.-X.; Li, C.; Liu, Y.-C.; Ma, Z.-Q.; Yu, H.-Y. Effect of Ti addition on high-temperature oxidation behavior of Co–Ni-based superalloy. J. Iron Steel Res. Int. 2020, 27, 1179–1189. [Google Scholar] [CrossRef]
- Zhou, P.; Gao, X.; Song, D.; Liu, Q.; Liu, Y.; Cheng, J. Role of Ru on the microstructure and property of novel Co–Ti–V Superalloy. J. Mater. Res. 2020, 35, 2737–2745. [Google Scholar] [CrossRef]
- Yang, H.-K.; Zhou, C.-Y.; Wang, H.; Yang, B. Phase equilibria in Ti-rich portion and thermodynamic re-optimization of Co-Ti system. J. Iron Steel Res. Int. 2022, 29, 914–924. [Google Scholar] [CrossRef]
- Wang, R.; Welsch, G. Evaluation of an experimental Ti-Co alloy for dental restorations. J. Biomed. Mater. Res. Part B Appl. Biomater. 2013, 101, 1419–1427. [Google Scholar] [CrossRef]
- Ruan, J.J.; Liu, X.J.; Yang, S.Y.; Xu, W.W.; Omori, T.; Yang, T.; Deng, B.; Jiang, H.X.; Wang, C.P.; Kainuma, R.; et al. Novel Co-Ti-V-base superalloys reinforced by L12-ordered γ′ phase. Intermetallics 2018, 92, 126–132. [Google Scholar] [CrossRef]
- Mutlu, I. Synthesis and characterization of Ti-Co alloy foam for biomedical applications. Trans. Nonferrous Met. Soc. China 2016, 26, 126–137. [Google Scholar] [CrossRef]
- Pang, X.Z.; Hu, J.; Zhan, Y.Z. Microstructural Characteristics of Ti-Co Alloys for Biomedical Applications. Adv. Mater. Res. 2013, 791–793, 469–473. [Google Scholar] [CrossRef]
- Liu, X.; Chen, S.; Tsoi, J.K.; Matinlinna, J.P. Binary titanium alloys as dental implant materials—A review. Regen. Biomater. 2017, 4, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yang, W.; Chen, G. On superplasticity of two phase alpha-titanium-intermetallic Ti–(Co, Ni)–Al alloy. Acta Metall. Mater. 1995, 43, 3571–3582. [Google Scholar] [CrossRef]
- Hu, K.; Huang, X.; Lu, J.; Liu, H.; Cai, G.; Jin, Z. Measurement of phase equilibria in Ti-Co-Pt ternary system. Calphad 2018, 60, 191–199. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhu, L.; Cai, G.; Liu, H.; Huang, J.; Jin, Z. Investigation of phase equilibria in the Ti-Co-Zr ternary system. Calphad 2017, 56, 260–269. [Google Scholar] [CrossRef]
- Fatoba, O.S.; Adesina, O.S.; Popoola, A.P.I. Evaluation of microstructure, microhardness, and electrochemical properties of laser-deposited Ti-Co coatings on Ti-6Al-4V Alloy. Int. J. Adv. Manuf. Technol. 2018, 97, 2341–2350. [Google Scholar] [CrossRef]
- Gromov, D.G.; Mochalov, A.I.; Pugachevich, V.P. CoSi2 formation in contact systems based on Ti-Co alloy with low cobalt content. Appl. Phys. A Mater. Sci. Process. 1995, 61, 565–567. [Google Scholar]
- Gromov, D.; Mochalov, A.; Pugachevich, V.; Kirilenko, E.; Trifonov, A. Study of phase separation in Ti-Co-N thin films on silicon substrate. Appl. Phys. A 1997, 64, 517–521. [Google Scholar] [CrossRef]
- Han, J.; Yoo, B.; Im, H.J.; Oh, C.-S.; Choi, P.-P. Microstructural evolution of the heat affected zone of a Co–Ti–W alloy upon laser cladding with a CoNiCrAlY coating. Mater. Charact. 2019, 158, 109998. [Google Scholar] [CrossRef]
- Yoo, B.; Im, H.J.; Seol, J.-B.; Choi, P.-P. On the microstructural evolution and partitioning behavior of L12-structured γ′-based Co-Ti-W alloys upon Cr and Al alloying. Intermetallics 2018, 104, 97–102. [Google Scholar] [CrossRef]
- Murray, J.L. The Co−Ti (Cobalt−Titanium) system. J. Phase Equilibria 1982, 3, 74–85. [Google Scholar] [CrossRef]
- Stein, F.; Merali, M.; Watermeyer, P. The Co–Ti system revisited: About the cubic-to-hexagonal Laves phase transformation and other controversial features of the phase diagram. Calphad 2019, 67, 101681. [Google Scholar] [CrossRef]
- Davydov, A.V.; Kattner, U.R.; Josell, D.; Waterstrat, R.M.; Boettinger, W.J.; Blendell, J.E.; Shapiro, A.J. Determination of the CoTi congruent melting point and thermodynamic reassessment of the Co-Ti system. Met. Mater. Trans. A 2001, 32, 2175–2186. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Wang, H. Microstructure and dry sliding wear resistance of CoTi intermetallic alloy. Intermetallics 2009, 17, 89–97. [Google Scholar] [CrossRef]
- Kaneno, Y.; Takasugi, T.; Hanada, S. Tensile property and fracture behavior of hot-rolled CoTi intermetallic compound. Mater. Sci. Eng. A 2001, 302, 215–221. [Google Scholar] [CrossRef]
- Wu, L.; Zeng, Y.; Pan, Y.; Du, Y.; Peng, Y.; Li, H.; Liu, S.; Zhang, L.; Liu, L. Thermodynamic description and simulation of solidification microstructure in the Co-Ti system. J. Chem. Thermodyn. 2019, 142, 105995. [Google Scholar] [CrossRef]
- Cacciamani, G.; Ferro, R.; Ansara, I.; Dupin, N. Thermodynamic modelling of the Co–Ti system. Intermetallics 2000, 8, 213–222. [Google Scholar] [CrossRef]
- Xi, S.; Chen, L.; Bao, L.; Han, J.; Yu, J.; Li, Z.; Xu, W.; Deng, B.; Wang, C.; Liu, X. Effects of alloying elements on the atomic structure, elastic and thermodynamic properties of L12-Co3(V, Ti) compound. Mater. Today Commun. 2021, 30, 102931. [Google Scholar] [CrossRef]
- Yasuda, H.; Takasugi, T.; Koiwa, M. Elastic Constants of Co3Ti and CoTi Intermetallic Compounds. Mater. Trans. JIM 1991, 32, 48–51. [Google Scholar] [CrossRef] [Green Version]
- Wollmershauser, J.; Neil, C.; Agnew, S. Mechanisms of Ductility in CoTi and CoZr B2 Intermetallics. Met. Mater. Trans. A 2009, 41, 1217–1229. [Google Scholar] [CrossRef]
- Jin, M.; Miao, N.; Zhao, W.; Zhou, J.; Du, Q.; Sun, Z. Structural stability and mechanical properties of Co3(Al, M) (M = Ti, V, Cr, Zr, Nb, Mo, Hf, Ta, W) compounds. Comput. Mater. Sci. 2018, 148, 27–37. [Google Scholar] [CrossRef]
- Xu, W.; Han, J.; Wang, Z.; Wang, C.; Wen, Y.; Liua, X.; Zhu, Z. Thermodynamic, structural and elastic properties of Co3X (X = Ti, Ta, W, V, Al) compounds from first-principles calculations. Intermetallics 2012, 32, 303–311. [Google Scholar] [CrossRef]
- Chaput, L.; Togo, A.; Tanaka, I.; Hug, G. Phonon-phonon interactions in transition metals. Phys. Rev. B 2011, 84, 094302. [Google Scholar] [CrossRef] [Green Version]
- Vítek, V. Intrinsic stacking faults in body-centred cubic crystals. Philos. Mag. 1968, 18, 773–786. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens. Matter. 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter. 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Chadi, D.J. Special points for Brillouin-zone integrations. Phys. Rev. B 1977, 16, 1746–1747. [Google Scholar] [CrossRef]
- Ikehata, H.; Nagasako, N.; Furuta, T.; Fukumoto, A.; Miwa, K.; Saito, T. First-principles calculations for development of low elastic modulus Ti alloys. Phys. Rev. B 2004, 70, 174113. [Google Scholar] [CrossRef]
- Guo, G.Y.; Wang, H.H. Gradient-corrected density functional calculation of elastic constants of Fe, Co and Ni in bcc, fcc and hcp structures. Chin. J. Phys. 2000, 38, 949–961. [Google Scholar]
- Liu, Y.; Chong, X.; Jiang, Y.; Zhou, R.; Feng, J. Mechanical properties and electronic structures of Fe-Al intermetallic. Phys. B Condens. Matter. 2017, 506, 1–11. [Google Scholar] [CrossRef]
- Wang, S.Q.; Ye, H.Q. Ab initioelastic constants for the lonsdaleite phases of C, Si and Ge. J. Phys. Condens. Matter. 2003, 15, 5307–5314. [Google Scholar] [CrossRef]
- Lu, W.; Li, C.; Yi, J.; Li, K. Stability and elastic properties of B2 CoX (X = Ti, Zr and Hf) intermetallic compounds as a function of pressure. Philos. Mag. 2017, 98, 203–218. [Google Scholar] [CrossRef]
- Wu, Z.; Zhao, E.; Xiang, H.; Hao, X.; Liu, X.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Patil, S.K.R.; Khare, S.V.; Tuttle, B.R.; Bording, J.K.; Kodambaka, S. Mechanical stability of possible structures of PtN investigated using first-principles calculations. Phys. Rev. B 2006, 73, 104118. [Google Scholar] [CrossRef] [Green Version]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Ozisik, H.; Deligoz, E.; Colakoglu, K.; Surucu, G. Structural and mechanical stability of rare-earth diborides. Chin. Phys. B 2013, 22, 046202. [Google Scholar] [CrossRef]
- Korozlu, N.; Colakoglu, K.; Deligoz, E.; Aydin, S. The elastic and mechanical properties of MB12 (M = Zr, Hf, Y, Lu) as a function of pressure. J. Alloy. Compd. 2013, 546, 157–164. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Duan, Y.; Huang, B.; Sun, Y.; Peng, M.; Zhou, S. Stability, elastic properties and electronic structures of the stable Zr–Al intermetallic compounds: A first-principles investigation. J. Alloy. Compd. 2013, 590, 50–60. [Google Scholar] [CrossRef]
- Jian, Y.; Huang, Z.; Xing, J.; Sun, L.; Liu, Y.; Gao, P. Phase stability, mechanical properties and electronic structures of Ti Al binary compounds by first principles calculations. Mater. Chem. Phys. 2018, 221, 311–321. [Google Scholar] [CrossRef]
- Feng, J.; Xiao, B.; Zhou, R.; Pan, W.; Clarke, D.R. Anisotropic elastic and thermal properties of the double perovskite slab–rock salt layer Ln2SrAl2O7 (Ln = La, Nd, Sm, Eu, Gd or Dy) natural superlattice structure. Acta Mater. 2012, 60, 3380–3392. [Google Scholar] [CrossRef]
- Tohei, T.; Kuwabara, A.; Oba, F.; Tanaka, I. Debye temperature and stiffness of carbon and boron nitride polymorphs from first principles calculations. Phys. Rev. B 2006, 73, 064304. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chávez, J.P. Controlling coexisting attractors of an impacting system via linear augmentation. Phys. D Nonlinear Phenom. 2017, 348, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal Elastic Anisotropy Index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [Green Version]
- Gaillac, R.; Pullumbi, P.; Coudert, F.-X. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter. 2016, 28, 275201. [Google Scholar] [CrossRef] [Green Version]
- Moreira, E.; Barboza, C.; Albuquerque, E.; Fulco, U.; Henriques, J.; Araújo, A. Vibrational and thermodynamic properties of orthorhombic CaSnO3 from DFT and DFPT calculations. J. Phys. Chem. Solids 2015, 77, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef] [Green Version]
- Kirihara, K.; Nagata, T.; Kimura, K.; Kato, K.; Takata, M.; Nishibori, E.; Sakata, M. Covalent bonds and their crucial effects on pseudogap formation in α−Al(Mn,Re)Si icosahedral quasicrystalline approximant. Phys. Rev. B 2003, 68, 014205. [Google Scholar] [CrossRef]
- LeClair, P.; Kohlhepp, J.T.; van de Vin, C.H.; Wieldraaijer, H.; Swagten, H.J.M.; de Jonge, W.J.M.; Davis, A.H.; MacLaren, J.M.; Moodera, J.S.; Jansen, R. Band Structure and Density of States Effects in Co-Based Magnetic Tunnel Junctions. Phys. Rev. Lett. 2002, 88, 107201. [Google Scholar] [CrossRef] [Green Version]
- Soulen, R.J.; Byers, J.M.; Osofsky, M.S.; Nadgorny, B.; Ambrose, T.; Cheng, S.F.; Broussard, P.R.; Tanaka, C.T.; Nowak, J.; Moodera, J.S.; et al. Measuring the Spin Polarization of a Metal with a Superconducting Point Contact. Science 1998, 282, 85–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Chong, X.; Jiang, Y.; Feng, J. Stability, electronic structure, mechanical and thermodynamic properties of Fe-Si binary compounds. J. Alloy. Compd. 2017, 693, 859–870. [Google Scholar] [CrossRef]
- Mulay, R.; Agnew, S. Hard slip mechanisms in B2 CoTi. Acta Mater. 2012, 60, 1784–1794. [Google Scholar] [CrossRef]
- Wang, J.; Sehitoglu, H. Dislocation slip and twinning in Ni-based L12 type alloys. Intermetallics 2014, 52, 20–31. [Google Scholar] [CrossRef]
- Wang, W.Y.; Xue, F.; Zhang, Y.; Shang, S.-L.; Wang, Y.; Darling, K.A.; Kecskes, L.J.; Li, J.; Hui, X.; Feng, Q.; et al. Atomic and electronic basis for solutes strengthened (010) anti-phase boundary of L12 Co3(Al, TM): A comprehensive first-principles study. Acta Mater. 2017, 145, 30–40. [Google Scholar] [CrossRef]
- Luo, W.; Kirchlechner, C.; Zavašnik, J.; Lu, W.; Dehm, G.; Stein, F. Crystal structure and composition dependence of mechanical properties of single-crystalline NbCo2 Laves phase. Acta Mater. 2020, 184, 151–163. [Google Scholar] [CrossRef]
- Yuan, F.; Li, G.; Liu, C.; Han, F.; Zhang, Y.; Muhammad, A.; Gu, H.; Guo, W.; Ren, J. Cross stacking faults in Zr(Fe,Cr)2 face-centered cubic Laves phase nanoparticle. Appl. Surf. Sci. 2020, 513, 145716. [Google Scholar] [CrossRef]
- Guénolé, J.; Mouhib, F.-Z.; Huber, L.; Grabowski, B.; Korte-Kerzel, S. Basal slip in Laves phases: The synchroshear dislocation. Scr. Mater. 2019, 166, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Shih, M.; Miao, J.; Mills, M.; Ghazisaeidi, M. Stacking fault energy in concentrated alloys. Nat. Commun. 2021, 12, 3590. [Google Scholar] [CrossRef] [PubMed]
- Vitek, V.; Paidar, V. Non-planar Dislocation Cores: A Ubiquitous Phenomenon Affecting Mechanical Properties of Crystalline Materials. Dislocations Solids 2008, 14, 439–514. [Google Scholar] [CrossRef]
- Im, H.J.; Lee, S.; Choi, W.S.; Makineni, S.K.; Raabe, D.; Ko, W.S.; Choi, P.P. Effects of Mo on the mechanical behavior of γ/γʹ-strengthened Co-Ti-based alloys. Acta Mater. 2020, 197, 69–80. [Google Scholar] [CrossRef]
- Russell, A.; Zhang, Z.; Lograsso, T.; Lo, C.; Pecharsky, A.; Morris, J.; Ye, Y.; Gschneidner, K.; Slager, A. Mechanical properties of single crystal YAg. Acta Mater. 2004, 52, 4033–4040. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Space Group | Strukturbericht Designation | Lattice Parameters | |||||
|---|---|---|---|---|---|---|---|---|
| a | b | c | ||||||
| -Ti | Im3m | A2 | 3.25 | 3.25 | 3.25 | 4.58 | −6.88 | 0 |
| 3.31 a | 3.31 a | 3.31 a | ||||||
| -Ti | P/mmc | A3 | 2.94 | 2.94 | 4.65 | 4.62 | −6.88 | 0 |
| 2.95 a | 2.95 a | 4.68 a | ||||||
| Ti2Co | Fd3m | 11.22 | 11.22 | 11.22 | 5.82 | −6.53 | −0.32 | |
| 11.30 b | 11.30 b | 11.30 b | ||||||
| TiCo | Pm3m | B2 | 2.92 | 2.92 | 2.92 | 6.71 | −6.45 | −0.39 |
| 2.99 b | 2.99 b | 2.99 b | ||||||
| TiCo2(c) | Fd3m | C15 | 6.63 | 6.63 | 6.63 | 7.52 | −6.20 | −0.29 |
| 6.72 b | 6.72 b | 6.72 b | ||||||
| TiCo2(h) | P/mmc | C36 | 4.70 | 4.70 | 15.29 | 7.48 | −6.21 | −0.29 |
| 4.73 b | 4.73 b | 15.43 b | ||||||
| TiCo3 | Pm3m | L12 | 3.60 | 3.60 | 3.60 | 7.95 | −6.07 | −0.27 |
| 3.61 b | 3.61 b | 3.61 b | ||||||
| 𝛼-Co | Fm3m | A1 | 3.52 | 3.52 | 3.52 | 8.97 | 0 | |
| 3.55 c | 3.55 c | 3.55 c | ||||||
| Phases | (GPa) | (GPa) | (GPa) | (GPa) | (GPa) |
|---|---|---|---|---|---|
| Ti2Co | 155.0 | 128.7 | 85.8 | ||
| TiCo | 220.1 | 137.9 | 70.3 | ||
| 203 a | 129 a | 68 a | |||
| TiCo2(c) | 280.0 | 135.1 | 100.4 | ||
| TiCo2(h) | 358.8 | 131.7 | 118.9 | 366.0 | 93.8 |
| TiCo3 | 236.2 | 149.1 | 113.7 | ||
| 228 a | 148 a | 129 a |
| Phase | (GPa) | (GPa) | (GPa) | (GPa) | ||
|---|---|---|---|---|---|---|
| Ti2Co | 137.5 | 41.7 | 113.7 | 0.362 | 3.294 | 1.397 |
| TiCo | 165.3 | 56.7 | 152.6 | 0.346 | 2.916 | 3.066 |
| TiCo2(c) | 183.4 | 88.1 | 227.8 | 0.293 | 2.082 | 8.649 |
| TiCo2(h) | 202.5 | 99.3 | 256.0 | 0.289 | 2.039 | 9.798 |
| TiCo3 | 178.1 | 77.4 | 202.8 | 0.310 | 2.302 | 6.601 |
| Phase | ||||
|---|---|---|---|---|
| Ti2Co | 366.4 | 3.016 | 5.760 | 2.678 |
| TiCo | 411.3 | 3.266 | 5.991 | 2.906 |
| TiCo2(c) | 494.0 | 3.820 | 6.325 | 3.423 |
| TiCo2(h) | 519.4 | 3.942 | 6.491 | 3.534 |
| TiCo3 | 388.0 | 3.285 | 5.600 | 2.937 |
| Phase | |||||
|---|---|---|---|---|---|
| Ti2Co | 5.5247 | 0.3596 | 6.5247 | 6.5247 | 6.5247 |
| TiCo | 1.7105 | 0.0348 | 1.7105 | 1.7105 | 1.7105 |
| TiCo2(c) | 1.3858 | 0.0127 | 1.3816 | 1.3816 | 1.3816 |
| TiCo2(h) | 0.8261 | 0.0064 | 0.7704 | 0.7704 | 0.8261 |
| TiCo3 | 2.6108 | 0.1066 | 2.6108 | 2.6108 | 2.6108 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, F.; Chen, M.; Wang, H.; Peng, H.; Li, B.; Huang, J. First-Principles Investigation on Phase Stability, Mechanical Properties, Bonding Characteristic and Slip Properties of Ti-Co Binary Intermetallic Compounds. Metals 2023, 13, 628. https://doi.org/10.3390/met13030628
Zeng F, Chen M, Wang H, Peng H, Li B, Huang J. First-Principles Investigation on Phase Stability, Mechanical Properties, Bonding Characteristic and Slip Properties of Ti-Co Binary Intermetallic Compounds. Metals. 2023; 13(3):628. https://doi.org/10.3390/met13030628
Chicago/Turabian StyleZeng, Fanlin, Mengjie Chen, Hongbo Wang, Hexiang Peng, Bei Li, and Jian Huang. 2023. "First-Principles Investigation on Phase Stability, Mechanical Properties, Bonding Characteristic and Slip Properties of Ti-Co Binary Intermetallic Compounds" Metals 13, no. 3: 628. https://doi.org/10.3390/met13030628
APA StyleZeng, F., Chen, M., Wang, H., Peng, H., Li, B., & Huang, J. (2023). First-Principles Investigation on Phase Stability, Mechanical Properties, Bonding Characteristic and Slip Properties of Ti-Co Binary Intermetallic Compounds. Metals, 13(3), 628. https://doi.org/10.3390/met13030628